Using Swabs for Cleaning Validation: A Review
By Douglas W. Cooper, Ph.D.
The Texwipe Company
Cleaning and cleaning validation are crucial to many industries. Clean swabs applied skillfully to areas that have been cleaned can be used to show the effectiveness of that cleaning. The swab, usually wet, is wiped across a cleaned region. The swab, or an extract from the swab, is then analyzed physically, chemically or biologically for contamination. The levels of contamination found are compared against the "blanks," the levels in swabs of the same type that were not rubbed on the surface being tested, and against a limit. Sufficient replication is done to make a statistically valid statement that cleaning was effective. The major choices - swabs, cleaning liquid, areas to be sampled, extraction, analytical method and statistical analysis - are discussed.
Introduction
Pharmaceutical, bio-medical device and even food preparation industries are concerned about the cleanliness - physical, chemical and especially biological - of their products. These industries have materials that need regular careful cleaning, and some have requirements for validating such cleaning, demonstrating that required levels of cleanliness have been met. They want to prove the efficacy of their cleaning methods. For example, "Cleaning and validation of cleaning are among the most critical issues facing producers of recombinant DNA protein products, monoclonal antibodies (MAbs), and oligonucleotide therapeutics," according to Adner and Sofer (1994). As noted below, the Food and Drug Administration (FDA) has taken validation of cleaning very seriously.
It is widely recognized that contamination control is crucial to these industries. Here we deal with the cleaning of surfaces, rather than the cleaning of gases or liquids, generally done by filtration. This article discusses the use of swabs (fabrics on handles or shafts) for cleaning validation.
Contaminants
Contamination comes from the environment, from the materials in use, the processes and the people. Viable and non-viable particles and various organic and inorganic materials often contaminate the surfaces of interest. Biological contamination in clean rooms, such as those used for aseptic filling, has been discussed in a monograph by the IES (Institute of Environmental Sciences, 1993).
Autoclaved pharmaceutical residues can be particularly hard to clean due to their proteinaceous nature, probably requiring an acidic cleaner, but only somewhat less hard are product residues, such as lipids, sugars and salts (Parenteral Drug Association (PDA), 1996), requiring different solvents. Solvents and cleaning compounds generally are relatively easy to remove, and product residues may or may not be.
Validation
Validation is proof or demonstration that something is what it claims to be. Carlberg (1995) defined it as "Full, detailed documentation that all processes and procedures are functioning in the manner they were designed for. Required by the FDA." Byers (in Carleton and Agalloco, 1985) gives several definitions, including, "A scientifically designed program to prove that a process consistently does what it is designed (intended) to do." The FDA (U.S. Food and Drug Administration, 1993) stated, "In the end, the test of any validation process is whether scientific data shows that the system consistently does as expected and produces a result that consistently meets predetermined specifications." The essence of all these definitions seems to be: documented, scientific proof of consistent successful performance.
The FDA Guide to Inspections (U.S. Food and Drug Administration, 1993), "intended to cover equipment cleaning for chemical residues only," includes:
- "FDA expects firms to have written procedures (SOPs) detailing the cleaning processes...."
- "FDA expects firms to have written general procedures on how cleaning processes will be validated."
- These procedures will "address who is responsible for performing and approving the validation study, the acceptance criteria, and when revalidation will be required."
- "FDA expects firms to conduct the validation studies in accordance with the protocols and to document the results of studies."
- Besides assuring chemical cleanliness, "the microbiological aspects of equipment cleaning should be considered. This consists largely of preventive measures...."
- Sterilization is not part of the scope of the document, except that reduction of the biological material will assist in successful sterilization and minimization of pyrogens.
- "Determine the specificity and sensitivity of the analytical method used to detect residuals."
- The sampling and analysis combination should be challenged to determine what fraction of the target material is actually sampled and then detected.
- " Direct sampling (e.g., with swabs) is "most desirable," although rinse sampling may be satisfactory.
References cited in this FDA document include Harder (1984), Smith (1992), and Fourman and Mullen (1993).
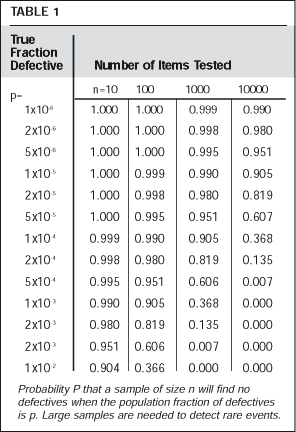
Recently, Gold (1996) presented an extended discussion of validation, stating, "Validation is necessary since testing of a product to assess its quality is not enough." Rare failures can escape testing but might be prevented by improved procedures. Table 1 shows the probability (binomial distribution) that a sample of size "n" will find no defectives, when the true fraction of the population is "p," which probability is (1-p) to the nth power. The table shows, for example, that if the true fraction defective is 1%, samples of size n=100 will find no defectives in 36.6% of the cases.
Sampling is clearly not enough. Validation requires checking the materials and equipment and procedures that constitute the process. The essence of validation is "establishing documented evidence that a process does what it purports to do." Gold (1996) emphasized that what is needed is routine, documented application of the scientific method. Documentation guidelines have been presented by Capote (1996).
Sterilization validation is a specialty with its own extensive literature (see Bruch in Morrissey and Phillips, 1993), and is not covered in detail here. Validation of Aseptic Pharmaceutical Processes by Carleton and Agalloco explores this subject in detail.
Cleaning Validation
Cleaning validation means proof that something has been cleaned, perhaps that the contamination levels have been reduced by at least a certain ratio or, more usefully, that they have been reduced below some target level. That means there will be some target limit level of surface contamination, and sampling and analysis will be carried out to assure that the area of interest almost certainly is cleaner than the target limit. Often, the locations hardest to clean will have to receive special attention to assure that they have been cleaned.
In an early paper on cleaning validation, Harder (1984) cited five crucial elements:
- A standard operating procedure (SOP) for cleaning (with a checklist)
- A procedure for determining cleanliness (e.g., rinse or swab, then analyze)
- An assay for testing for residual drug levels
- Pre-set chemical and microbial limit to which the equipment must be cleaned
- A protocol for cleaning validation
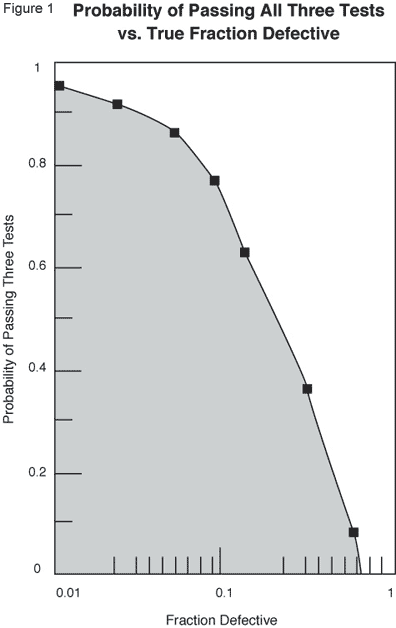
Harder recommended that the procedure be tested by requiring it to be successful on three successive cleanings. (See Figure 1) and that there should be periodic revalidation, as well as revalidation after significant changes.
Jenkins and Vanderwielen (1994) presented an overview of cleaning validation, covering strategy, determination of residue limits, methods of sampling, and analysis. They noted that "increased use of multipurpose equipment" has produced increased interest in cleaning validation. The cleaning protocol must be thorough and must be checked. Training is essential. A validation program requires:
- Criteria for acceptance after cleaning
- Appropriate methods of sampling
- A maximum limit set for the residues
- The test methods must themselves be tested
Products to be tested may best be put into groups, rather than testing all of them. The most important may not be the highest-volume products, but those capable of causing the largest possible problems if contaminated or if they contaminate other products. (Solubility of the drug is an important issue.) Equipment may also be tested as groups.
Geigert et al. (1994) emphasized the importance of cleaning validation, as making good business sense and being required by cGMP.
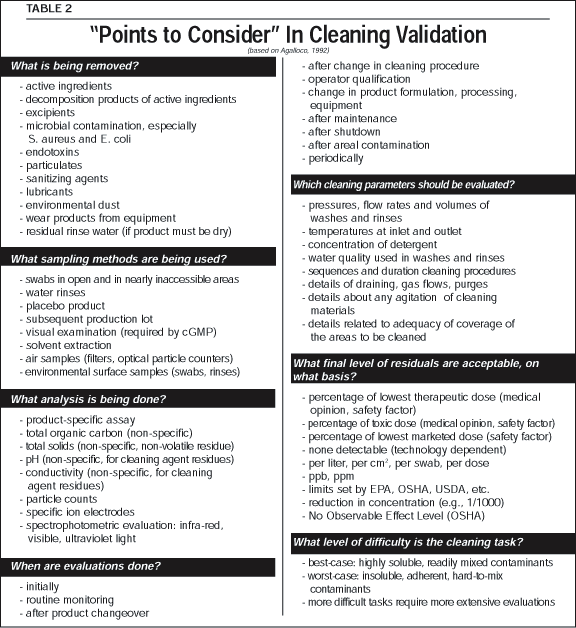
Table 2 has "points to consider" for cleaning validation, summarizing an excellent article on the topic by Agalloco (1992), who stressed that not all of these points need equal weight.
Recently a highly useful book has been published on cleaning and cleaning validation (PDA, 1996), covering process equipment design, cleaning mechanisms, automatic cleaning equipment, validation studies, sampling methods, analytical methods, validation acceptance criteria and special considerations for multi-product facilities. The following sampling methods provide various levels of assurance concerning cleaning:
- Visual inspection
- Easy
- Qualitative
- Subjective
- Easy
- Rinse water sampling and analysis.
- Sampling is easy
- Analysis can be quantitative, using pH, conductivity, particle count, microbial count, total organic carbon determination, spectrophotometry, bioassays, limulus amebocyte lysate for pyrogens
- Recovery factor is uncertain; it involves dilution.
- Sampling is easy
- Surface sampling and analysis
- Removes adherent materials
- Analysis can be quantitative
- Precise definition of the area sampled
- Removes adherent materials
- Surface sampling from coupons
- Quantitative
- Depends on whether coupons are equivalent to the surface of interest
- Requires removing coupons from system...
- Quantitative