Risk analysis
Here you will find answers to the following questions:
|
1 Development of the risk analysis
For a long time, risk analyses have been being carried out in the chemical-pharmaceutical industry in the field of safety engineering. They started in the early 60s when the aerospace industry developed risk analyses. Ultimately, technical systems had to be realised which had to be 100% operational, far from any maintenance unit. Breakdowns would have (and did, as history teaches us) brought unexpected consequences. In any case, human lives were put at stake. So, already in the development phase of a product (Apollo rocket), every possible type of malfunction had to be ruled out. To do this, methods with which the aggregates could be systematically analysed for their failure rate, were developed. This was the hour of birth of Failure Mode and Effects Analysis (FMEA).
For a long time, such analyses were only used in high-risk industries (aerospace, nuclear engineering, etc.). In the context of the emerging quality instruments in the 80s, methods had to be found to help increase the quality of industrially created products. These methods made a crucial breakthrough in the automotive and semiconductor industry and for their suppliers in the 80s and 90s. The cost pressure on these industry branches increased drastically and the industry was required to simultaneously increase quality and reduce costs. The use of risk analyses was a question of survival and the ability to reach the target of high quality with low costs was of crucial importance.
|

In the pharmaceutical industry, the discussion of risk analyses often focuses on the costs. Yet risk analyses were developed not to increase costs, but to save costs. These methods should therefore also be implemented in this spirit.
Experience shows that early system analysis can lead to significant advantages for investment projects. The form of risk analysis used is unimportant. What is crucial is an awareness of the GMP risks associated with the selection of certain machines, apparatus and equipment. It should be a matter of course that, at the time of ordering a technical system, the requirements of the future user should be present. These user requirement specifications form an essential basis for the execution of a risk analysis.
| Example |
Basis |
Methods |
|---|---|---|
Definition of GMP requirements for a process facility |
Experience with other facilities, GMP guidelines, laws |
Structured procedure, company-specific risk analysis |
Construction review of process facility (fill line, blender, etc.) |
User requirements, technical specification, construction documents |
FMEA, company-specific risk analysis |
Definition of GMP requirements for old facilities |
Experience with the facility, facility documentation, GMP guidelines, laws |
FMEA, company-specific risk analysis |
Risk analyses can be applied in various task areas. It is always important to be in a position where the changes can still be carried out cost-effectively, but where important basic conditions have already been established. Some possible uses for risk analyses are listed in figure 19.B-3.
| Examples of risk analyses |
||
|---|---|---|
| Area of application |
Time of the risk analysis |
Content of the risk analysis |
Validation master plan |
Rough plan available |
What technical facilities and systems are to be classified as GMP-critical, and which as uncritical? The influence of the systems on the product quality is crucial. |
Qualification |
Conclusion of engineer planning; Design review; Construction review |
GMP-critical systems are inspected in detail for compliance with the GMP requirements and for whether or not GMP risks have been take into account adequately in the design. |
Computer system |
Conclusion of the specification (functional and technical specification) |
Review of the extent to which the specification contains GMP risks that must be tested (validated) in later project phases. |
Process validation |
Conclusion of process development |
What GMP risks are included in the sequence of operation? In what processing step of the industrial implementation are GMP problems to be expected? |
Cleaning validation |
After development of the cleaning process |
What GMP risks are contained in the processing steps? Which are worst-case products for the established cleaning sequence? |
and others |
||
Official requirements for risk analyses
The monitoring by the authorities also places increasing value on the execution of risk analyses. Information on this is given in particular in the ICH Q9 Guideline "Quality Risk Managment" and in Annex 15 of the EU GMP Guideline.
The following methods are interesting for use in the pharmaceutical industry:
- Failure Mode and Effects Analysis (FMEA)
- Fault Tree Analysis (FTA)
- Ishikawa method (Fishbone analysis)
- HACCP
The ICH Q9 defines risk analysis as:
"The estimation of the risk associated with the identified hazards."
Annex 15 defines risk analysis as follows:
"Methods to assess and characterise the critical parameters in the functionality of an equipment or process."
Both definitions show that risk analysis is a procedure with which critical parameters should be determined or evaluated.
Annex 15 also goes into the increasing importance of risk analysis where changes have been made:
"All changes that may affect product quality or reproducibility of the process should be formally requested, documented and accepted. The likely impact of the change of facilities, systems and equipment on the product should be evaluated, including risk analysis. The need for, and the extent of, requalification and re-validation should be determined." (Annex 15, paragraph 44)
The ICH Q9 deals with the different forms of risk analysis in much more detail and also introduces these briefly.
With the help of a documented risk analysis, it is possible to show why certain facility functions or procedure steps should be classified as critical or non-critical. This evaluation is then a traceable basis for qualification and validation activities.
Type of risk consideration
The type of risk analysis that is suitable for the project must be decided within the project team. This decision can only be reached within the project. For other parties, it is irrelevant if this is a formal procedure according to existing methods or a newly developed approach. What is crucial is whether or not an outsider can trace how it was applied and if it can be clearly explained. Possible risk analyses are, for example, Failure Mode and Effects Analysis (FMEA), Fault Tree Analysis, Fishbone diagram (Ishikawa diagram), HACCP, free form of risk analysis.
2 FMEA - Failure Mode and Effects Analysis
2.1 Development
FMEA is a method that was developed by NASA during the Apollo program. Through various phases of further development, it has experienced significant developments through application in the mass industry of automobile and machine construction and in the semiconductor and microelectronics industry. The practical feasibility of the method was proven by these applications. FMEA is allegedly the most used form of risk analysis.
FMEA is a quantitative risk analysis, the result of which is a list of risks with an assigned risk value.
To date, every industry has changed certain aspects of FMEA in implementation. The introduction of FMEA in the operational sequence of pharmaceutical companies also requires adjustments. In recent years, especially in the evaluation system, a certain "pharmaceutical standard" has been established which must be taken into account in the procedure described below.
2.2 Procedure during FMEA
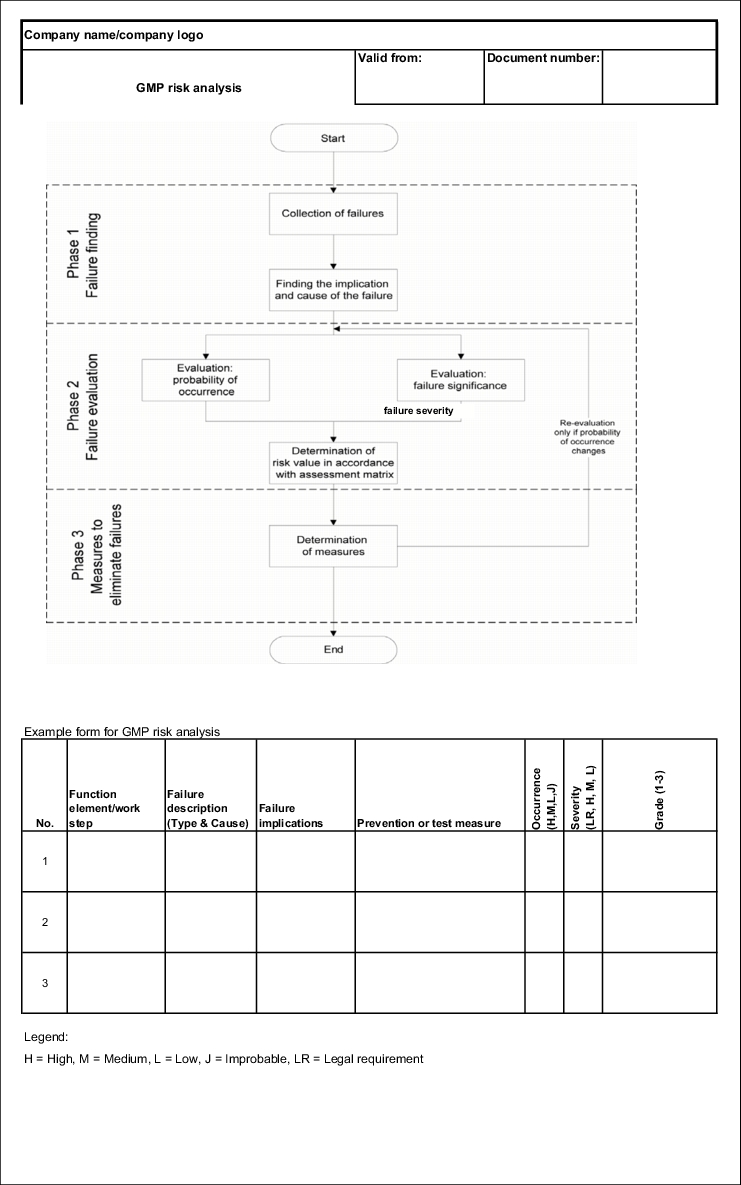
Figure 19.B-4 shows a flow chart of the sequence of a GMP-FMEA. Three important stages can be distinguished:
- Failure finding
- Failure analysis
- Measures to eliminate failures
Here, a failure is to be understood as a GMP risk. Thus, for example, a failure can be a leaky cable duct through a wall in a clean room or a difficult to clean point in a coater or a ball valve in the distribution ring of a WFI system.
|

2.3 Failure finding
The first phase of FMEA is to find GMP deficits. Before GMP risks can be evaluated, they must first be identified and described.
If the FMEA is only used for a rough evaluation of systems, then the team discusses the entire system and describes general GMP deficits that could be caused by the system. At this level, it is usually not necessary to deal with the cause of failure, as this can often be very multifaceted (e.g. see figure 19.B-5).
| System |
Failure |
Cause of failure |
Failure implications |
|---|---|---|---|
Water for injection (WFI) |
Microbial contamination of the WFI |
various |
Microbial contamination of products |
If the FMEA is for subsystems, failure finding is carried out in significantly more detail. Individual subsystems are examined and the causes of failure and failure implications are investigated, so that later evaluation is easier.
| System |
Failure |
Cause of failure |
Failure implications |
|---|---|---|---|
Water for injection (WFI), distribution system |
3-D rule not complied with |
Faulty execution |
Microbial contamination of products |
Washing system for sterile primary containers |
Exhaust air air contains particles/organisms |
No sterile filter planned |
Microbial contamination of products and high content of particles |
The most time-consuming part of the FMEA is the failure finding. All systems or subsystems must be checked for their possible failures. Even failures that may appear hypothetical must be taken into account, as practice shows that every failure occurs at some time.
Failure finding, in particular in the initial phase of execution of FMEAs, is time-consuming. If several similar systems have already been analysed, then there will be many similarities also in the occurrence of failures, which reduces the effort for this phase. Generally, failures are found by the team members who work with the corresponding facility in practice and through the application of creativity methods (e.g. brainstorming, brainwriting, etc.), which are not dealt with in detail here.
At this time, it is important to take into account all GMP-relevant aspects and official requirements. Official requirements which, if not complied with, can also be classified as failures, can be derived from the bodies of rules (e.g. CFR, EU GMP Guideline, Guides, Guidelines, etc.). However, it is also important to take into account the state-of-the-art, as one of the official requirements is to consider the state-of-the-art in the design of technical systems. Determining the state-of-the-art is not always easy. The IPSE Baseline® Series documents are helpful. These documents show how today's technical systems should be designed by pharmaceutical production companies. As these documents are compiled by engineers and are agreed with the FDA, they are good guidelines for the state-of-the-art.
If all feasible failures have been documented with cause of failure and implications of failure, these must be put into some order. Different causes of failure often lead to the same failures and the same failures have different implications. This too is documented in a traceable manner using the form (see figure 19.B-7).
|
|


2.4 Failure evaluation
In the failure evaluation phase, principly only one line in the FMEA form is evaluated at a time. If, for example, there are several causes of failure which are linked with a failure in separate lines, these must be taken into account separately. However, if the different causes of failure were grouped together in one line, the causes of failure are evaluated together.
In general, the following aspects of failures are evaluated:
- Probability of occurrence of the failure (O)
- Severity of the failure (S)
- Probability of detecting the failure (D)
These three defect characteristics are assigned numerical values from which the risk priority number is determined by multiplying the three values with each other (see figure 19.B-8).
| Calculating the risk priority number (RPN) |
|---|
RPN = O x S x D O = Probability of occurrence, S = Severity, D = Probability of detection |
It is of fundamental importance that the three evaluations take place independently of each other. The probability of occurrence or the probability of detection of a failure must not be included in the severity of the failure. For example, if the implications of a failure lead to S = 5, then the number must not be reduced purely because this failure only occurs once a year. This step is carried out in the concluding calculation of the risk priority number RPN (see figure 19.B-8).
The scale for the numerical values can be defined by the company applying them. So, for example, to achieve a rough classification of systems, it may be advisable to make an evaluation of 1 to 3 in each case. This way a result can be reached more quickly and efficiently. The evaluation aids described in figure 19.B-9 to figure 19.B-11 are a suggestion. Each company must develop evaluation aids when introducing FMEA.
Probability of occurrence (O)
For a GMP risk, the frequency with which a failure occurs or can occur is of great importance. The more frequently a failure occurs, the higher the risk. Unlike in other industry branches, evaluation in the pharmaceutical industry is usually carried out with number values between 1 and 5. O = 1 means rare occurrence and O = 5 means frequent occurrence (see example in figure 19.B-9). Other industry branches use a system of 1-10 and thus offer more detailed classification. This is not necessary for the current application in the pharmaceutical industry and also saves valuable time in the FMEA meetings, as it is easier to reach an agreement when the margin is narrower.
| Example of an evaluation aid for the probability of occurrence |
|
|---|---|
| Probability of occurrence |
Evaluation |
Improbable: or It is improbable that a failure will occur. |
1 |
Very low: or The construction generally corresponds to previous drafts in which few failures occurred (max. 3 times per year). |
2 |
Low: or The construction generally corresponds to previous drafts in which failures occurred occasionally (max. once per month). |
3 |
Moderate: or No information is available |
4 |
High: or It is almost certain that failures will occur to a large extent. |
5 |
Failure severity (S)
The severity of a failure is fundamental for its assessment. The severity is usually determined by the implications of the failure. If, for example, a patient is injured as a result of a failure, the severity must be classified as high. On the other hand, if only a review of the technical system is required, the severity is to be classified as low. The numerical values from 1 to 5 ascend with increasing severity (see figure 19.B-10).
| Example of an evaluation aid for the failure severity |
|
|---|---|
| Severity in terms of product quality |
Evaluation |
No disadvantageous impacts on the product quality can be deduced. |
1 |
The failure has an impact on the technical sequences, but there is no impact on the product quality |
2 |
Slight deviations in the product specifications can occur which require moderate measures (e.g. higher monitoring frequency in final testing, additional testing, etc.). |
3 |
Significant deviations in the product quality can occur which require extensive measures (e.g. rejection of a batch, recall of products, etc. ). |
4 |
Deviations can occur that might cause injury to consumers. |
5 |
Probability of detection (D)
To determine the GMP risk, it is important to know if a failure that has occurred will be detected or if it will only be noticed once it has reached the customer. This also includes a failure that is detected in the pharmacy, as this too is a customer.
The easier it is to detect the failure, the lower the risk. Therefore, the numerical value falls from 5 to 1 the higher the probability of detection. D = 1 is thus a value that can only be achieved if a fully automatic 100%-control is integrated in the production sequence. D = 5 means that a failure is not detected (see figure 19.B-11)
When evaluating the probability of detection, the test measures that are already planned or designed at the time of executing the FMEA must be taken into account. This being the case, they must also be documented in the FMEA form in connection with the evaluation.
| Example of an evaluation aid for the probability of detection |
|
|---|---|
| Probability of detection |
Evaluation |
High: or Operating failure that is almost certainly noticed in the subsequent operations. |
1 |
Average: or Obvious failure that is very probably detected in the subsequent operations. |
2 |
Low: or Easy to recognise failure, which is controlled. |
3 |
Very low: or Defect characteristic that is difficult to recognise |
4 |
Improbable: |
5 |
Evaluation of the risk priority number (RPN)
The risk priority number (RPN) is determined by multiplying values O, S and D.
This results in an RPN of between 1 and 125 with the above-mentioned assessment values. In addition to the RPN, the company's risk tolerance is of great importance. That is, where the limit is set by the company for the determination of measures. It is necessary to clarify the RPN from which measures must be initiated in order to reduce the risk. It must also be clarified whether this should be a single limit or if there should be two limits, for example (see figure 19.B-12).
|

If the set limits are exceeded, the FMEA team must define measures that will reduce the RPN.
2.5 Measures to eliminate failures
The definition of measures to be implemented when the RPN is exceeded is a preventative failure prevention program which is very cost/benefit efficient from a very early stage. Below are some examples of such measures:
- Introduction of IQ or OQ tests
- Introduction of an additional quality check
- Changes to the facility to completely prevent certain failures
- Introduction of an additional point in the context of preventative maintenance
When defining measures, their impact on the RPN and the previous evaluation of the individual criteria, O, S and D are crucial. It is therefore advisable to carry out a second evaluation after the measures have been established.
If you consider the above-mentioned examples of measures, one of the results of a detailed FMEA could be a complete list of all necessary IQ and OQ or PQ tests. This means that this information is available from a very early stage and the course of the project or the necessary resources can be planned more accurately. In addition, time-consuming agreements at a later time are no longer necessary which, according to experience, weigh heavily on the project if the necessary tests have not been defined at such an early stage.
3 Introduction of a GMP risk analysis according
to FMEA method
The introduction of a GMP risk analysis according to the FMEA method must be very well planned. Figure 19.B-13 shows a checklist of all the important points, listed in chronological order, which should be carried out during introduction of FMEA. It is the task of the project planning for the implementation of a GMP-FMEA to decide the timeframe in which the individual points are to be processed.
| Checklist for the introduction of a GMP risk analysis using the FMEA method |
|
|---|---|
Project start |
|
Appointment of project manager |
The project manager must have the corresponding authorities |
Establishment of the project team |
The project team must include employees representing production, engineering and quality assurance. Further project members must be selected according to the project demand. |
Establishment of the fields of consideration |
This is where the project scope is defined, i.e. which systems are to be investigated. |
Establishment of the depth of consideration |
Is it a rough analysis or a detailed analysis? |
Establishment of the classification |
Will aspects O, S and D be assessed with values from 1 to 5 or is another system being used? |
Evaluation aids for determining; O, S and D must be compiled |
These aids should ensure that different teams can achieve equivalent results. |
Development of principles for the GMP risk assessment |
Efficient processing of the GMP risk analysis is facilitated through the existence of operational and official requirements and the state-of-the-art. |
Establishment of RPN limits |
Have one or two limits been established and how high are they? What happens if the limits are exceeded? |
Establishment of the FMEA teams |
These will not always agree with the project team, in particular if the projects are extensive. |
Training of moderators |
At least one FMEA team member should be trained as a moderator. |
Training of the FMEA team members |
The FMEA should be trained using examples. |
Execution of GMP risk analysis in first teams |
Experiences in the first teams should be particularly well documented. |
Implementation of the decided measures |
The measures must be implemented, otherwise the risk assessment is meaningless |
Monitoring of the measures |
Likewise, the implementation of the measures must be monitored. |
3.1 Advantages
As a method, FMEA has many advantages which, in other industry areas, have ultimately lead to FMEA asserting itself significantly more strongly over other risk assessments (see figure 19.B-14.)
| Advantages of FMEA |
|---|
|
Quantitative evaluation of GMP risks
Quantitative evaluation of GMP risks is possible with FMEA. In the form shown below, this results in a risk value (risk priority number) between 1 and 125. The risk increases as the value increases. The company or team can decide for itself where the tolerable risk, and thus a limit, is set (see figure 19.B-12). Usually, this is a risk value between 15 and 30. The risk priority number (RPN) means that it is possible to compare risks from different technical systems. This is of particular significance for strictly cost-limited projects.
Ranking of GMP risks
By determining an RPN for each GMP risk, it is possible to compile ranking lists. This distinctly simplifies priority determination. The decision regarding which aspects must be handled with priority is documented in a traceable manner. For projects with a limited budget, an efficient instrument is available for implementing the available resources efficiently for the critical GMP risks.
Qualification aspects are defined at an early stage
Since the detection of GMP failures (probability of detection) is an evaluation criterion within FMEA, the tests of IQ, OQ and PQ can be established at a very early stage. This is very advantageous for resource planning for qualification.
Different levels of consideration
Depending on the project progress, FMEA can be executed in more or less detail. This means that the critical systems can be classified in a rough planning stage and, in a later step, the critical systems can be checked at aggregate level for GMP-critical facility parts and functions. The results will be correspondingly rough or detailed. Thus, every company can adapt FMEA to its requirements.
Team approach
The composition of the group that carries out the FMEA is crucial for the success of the FMEA. An optimal result is achieved, as people with different levels of knowledge are collaborating in the group. In order to achieve this, the group must work out the result together, with the later user and engineering always being represented in the group.
Comprehensive method
In contrast to other risk analysis methods, FMEA can also be used to carry out other risk analyses, e.g. safety, environmental protection, technical risk analysis. This would make it possible to investigate and consider all risks with this method.
3.2 Disadvantages
The disadvantages of FMEA are essentially rooted in its initial implementation, but also in the continuity generally required for carrying out a risk analysis.
| Disadvantages of FMEA |
|---|
|
Training
Before carrying out a FMEA, the participants must be trained in its application. This is indispensable as the participants in the group will otherwise be working with different perceptions and requirements. This leads to endless discussions and dissatisfaction in the group members and during the FMEA meetings.
For this reason, a FMEA training course should be attended, in which the team members are familiarised with the method and gain certainty in how to execute a FMEA. If all members of a FMEA know what they are dealing with, FMEA meetings can be held efficiently. A training course of 0.5 - 1.5 days should be held, depending on the level of knowledge of the employees.
In addition to the FMEA training, one member of each FMEA team should be trained as a moderator. The moderator has the task of guiding the team to a result during the meetings. This does not mean that the moderator develops the solution, but that he helps the team to reach the target. Therefore, the moderator must know the method inside out and must also be able to explain it. He must always have the objective of the FMEA in sight. Often, in cases of dispute within the team, the moderator provides a consensus.
Implementation
The initial implementation of FMEA in the company should not be rushed. The staff must first become familiarised with the method and practice it for a long period of time before each individual can form an actual image of the effectiveness of the method. This process takes time. Unfortunately time is rarely available in the company.
If the implementation phase is too short, this often leads to dissatisfaction of the executives and staff. The former are disappointed by the results and the latter are overtaxed with expectations. The implementation phase can be shorter if the staff and teams are provided with corresponding aids, e.g. in the form of coaching or training.
Resource availability
In order to be able to hold FMEA meetings efficiently, all members must be able to participate in the discussion. If this is not possible, it inevitably leads to losses of efficiency and the result suffers from the lack of resource availability. Therefore, FMEA meetings must be prepared in good time and all team members must be aware of the associated priority.
Quasi-objectivity of the RPN
As the RPN (risk priority number) is a numerical value, you could incorrectly assume that it is an objective estimate of the facts. However, the RPN is only an indicator of the significance of a GMP risk. It does not matter if the RPN is 15 or 16, but rather if a value is 15 or 30. You must always be aware that the RPN is the result of a subjective estimate by the team. Just because the process for forming an opinion ends in a tangible numerical value does not make it more objective, but rather it remains a subjective estimate.
This would mean that different teams could easily obtain different results. All those involved must be aware of this fact in order to avoid misinterpretations.
An example operating procedure for carrying out risk analysis according to the FMEA method is shown in figure 19.B-16.
|
|
|
|
|
|
|
|








4 Company-specific risk analysis
The fact that a GMP risk assessment was carried out according to a current method is not crucial for its efficiency. Rather, what is crucial is whether or not the risk analysis is anchored in the project or in the company and if it is suitable for showing the GMP risks transparently and for documenting them.
Therefore, the "company-specific risk analysis" is attributed a high level of importance. Different models of highly simplified risk analyses are often practised, which supply less detailed results but can be processed in a shorter period of time.
4.1 Advantages
The advantages of company-specific risk analysis are essentially its better adaptability and thus the higher level of acceptance within the company. In addition, a resource-saving route is often chosen.
| Advantages over an existing method |
|---|
|
Risk analysis is adapted to the company requirements
Company-specific risk analysis is ideally adapted to the requirements of the company. Therefore, this form can be the best instrument for the respective company. The requirements of different departments can be accounted for to a greater extent or the human resources requirements can be more easily taken into account.
High level of acceptance
Since the method is developed in and with the company, a company-specific risk analysis usually has a higher level of acceptance than an "external" method. This is of great importance for the introduction of a GMP risk analysis, as it can decide on the success or failure of the implementation.
Resources saved
With company-specific risk analysis, methods can also be established that are adapted to the company's human resources.
4.2 Disadvantages
Company-specific risk analysis also has disadvantages compared with existing methods (see figure 19.B-18)
| Disadvantages compared with an existing method |
|---|
|
In-house development costs time and resources
It is easily comprehensible that it is more costly to develop a GMP risk analysis in-house than to adopt a tried and tested method. In any case, this applies to the development stage. This additional effort, however, can be justified if corresponding resources can be saved in the subsequent application time. The importance of time must not be underestimated. A method must be developed but also tested. If inadequacies come to light in the testing stage, they can be eliminated in the appropriate manner. However, if a method has to prove itself immediately in practice, e.g. because a new project is at hand, then there is the risk that there will be not time for testing and deficiencies will first occur at a later stage when it is too late to make cost-effective corrections.
Specialist knowledge and experience
If nobody within the company has operational experience of risk analyses, it is advisable to involve a specialist in the project team, or someone with corresponding experience. Many possibilities that appear plausible in theory are much less suitable in practice. Only if the design of risk analyses is derived from practical experience (this implies corresponding loops), does the project have the prospect of success. If this point is neglected, methods are often developed which come up against reservations, criticism or opposition from the employees who are supposed to implement the methods. The damage that can result from the employees not adequately identifying with procedures or rejecting them out and out, can barely be quantified, but is significant in any case. Specialist knowledge and experience in dealing with risk analyses need not be taken into account to such a great extent for the use of standardised risk analyses, but also in this case the acceptance of the method is crucial.
Exchange throughout the company is difficult
For the design, improvement and further development of company-specific risk analyses, a smaller public is available, in contrast to the tried and tested methods. This means that most suggestions have to come from within the company. Known methods, such as FMEA, on the other hand, are often publicly discussed and opinions and experiences are exchanged. Thus, more widespread methods can be optimised more quickly and practice oriented.
4.3 Procedure
On the basis of the above-mentioned points, the existing risk analyses should first be intensively investigated in the company. If no risk analysis is found that meets the internal requirements, then the development of a company-specific risk analysis is on the cards.
However, the last resort should be doing this entirely independently. Many companies will be more than happy to provide you with experiences they have made. Therefore, the best path to take is to first ask other companies about their experiences and the methods they use. If this too brings you no further, the last resort is to develop a method alone or in collaboration with another company.
An in-house development has a greater chance of acceptance if it is based on an existing method. So, for example, FMEA can be used as a basis and converted accordingly, as shown in the following example.
4.4 Example
FMEA should be used as a basis for this example. After investigating FMEA, the following procedures should be simplified:
- Evaluation of the risk not to be carried out with numbers
- Only two evaluation aspects are to be evaluated (three was too many)
- A simple assessment grid should give clearer requirements for the final assessment.
- The method should be more simple and transparent than FMEA
Modifications compared with FMEA
The evaluation of the risk should not be carried out with numbers, but with a descriptive grading. This will make it clearer that the value is not objective. There should be evaluation aids for these gradings, as in the FMEA, which facilitate the evaluation of risks.
The three evaluation aspects should be reduced to two. The evaluation should focus on the probability of occurrence and the severity of a failure (see figure 19.B-19). The basic consideration is that the probability of detection is also derived from the measures that are to be decided on for certain risks.
The probability of occurrence is graded as follows:
- Improbable
- Low
- Average
- High
The severity is graded as follows:
- Low
- Average
- High
- Legal and quasi-legal requirement
Due to the fact that no numerical values are determined, no risk priority number can be formed. However, there is a clear matrix, with the help of which a risk value can be determined and the necessary measures can quickly be deduced.
| Severity |
|||||
|---|---|---|---|---|---|
| Low |
Average |
High |
Legal Req. |
||
Occurrence |
High |
1 |
2 |
3 |
3 |
Average |
1 |
1 |
2/3 |
3 |
|
Low |
0 |
0 |
2 |
2 |
|
Improbable |
0 |
0 |
0 |
1/2 |
|
Legend 0: These fields do not require any GMP measures 1: These aspects must be reviewed during qualification 2: These aspects must be reviewed during qualification, where possible also with function tests, and measures may have to be taken to reduce the probability of occurrence (change of facilities). The process may have to be supplemented with quality checks to increase the detection of failures. 3: These aspects must be reviewed during qualification, corresponding measures for reducing the probability of occurrence must be initiated or the detection of failures must be guaranteed through adequate checks. |
|||||
If, for example, a moderate failure is found, the function must be checked during qualification, depending on the severity of the failure. It may even be necessary to change the design of the facilities or to incorporate additional quality checks in the production process.
With this modification of the FMEA method, the procedure before evaluation remains the same. This means that the potential failures and risks must first be found. The changes result in the procedure shown in figure 19.B-20.
|

There is also a significant difference in the fact that the loops now only relate to the reduction of the probability of occurrence. Other facility and process changes (such as additional quality controls) can no longer be included in the assessment matrix due to the change of method (see figure 19.B-20).
The form for this type of risk analysis can be seen in figure 19.B-21.
|
|


An example operating procedure for carrying out company-specific risk analysis is shown in figure 19.B-22.
|
|
|
|



5 Hazard Analysis of Critical Control Points
HACCP (Hazard Analysis of Critical Control Points) was developed in the early 60s as part of the NASA program, so that food, which was 100% safe against contamination by bacteria, pathogenic viruses, toxins or chemical and physical hazards, could be produced for the space program. HACCP replaced the end-product testing and at the same time guaranteed the safety of food. In the meantime, HACCP has now gained recognition world-wide as an efficient tool for quality assurance in the production of foodstuffs.
HACCP focuses on hygiene, which is of crucial interest for any plant that processes food. The legislator too has acknowledged the significance of hygiene for this area. An important milestone for this method was the legal obligation of food processing plants to execute this method (EC Directive 93/43, Hygiene of Foodstuff). Through the requirements of § 3 of the Hygiene Regulation, this form of risk analysis became standard in the industry.
In accordance with article 3(2) of the EC Directive 93/43 Hygiene of Foodstuff, the concept of HACCP should satisfy the following principles:
- Analysis of the potential risks for food in the processes of a food company;
- Identification of the points in these processes at which risks could occur for food;
- Definition of which of these points are critical for food safety - the "critical points";
- Establishment and execution of effective test and monitoring processes for these critical points, and
- Review of the risk analysis for food, the critical control points and the test and monitoring processes in regular intervals and each time the processes are changed in the food company.
The pharmaceutical industry also became aware of this form of risk analysis as it became more widespread. As the authority also responsible for food, the FDA had already taken a leading role in introducing HACCP for food processing. However, as hygiene also plays an important role in pharmaceutical production, it is worth considering this form of risk analysis here too.
Procedure of HACCP
In HACCP, the team approach has also proven its worth and prevailed. This means that as many task areas as possible should be represented in the team, e.g. quality assurance, engineering, laboratory and production. A clear and uniform definition of terms is also important. The following key terms are of importance for HACCP (source: HACCP Guidelines, FDA):
Hazard:
"Hazard [...] means a biological, chemical, or physical property that may cause an unacceptable consumer health risk." (HACCP Guideline, FDA, 1997)
Hazard is an event or circumstance that can influence a product in such a way that may cause a direct consumer health risk. The hazard can be of a biological, physical or chemical nature.
|

Risk:
"Risk means an estimate of the likely occurrence of a hazard." (HACCP Guideline, FDA, 1997).
Critical control point (CCP):
"Critical control point [...] means a point at which loss of control may result in an unacceptable health risk." (HACCP Guideline, FDA, 1997)
A critical point is therefore a stage, step or phase, at which a detected hazard is to be eliminated through targeted and controlled measures or reduced to an acceptable level with the clear aim of controlling the hazard.
The general procedure for HACCP is the same as for all risk analyses:
- Search for possible failures/problem points
- Evaluation of these problem points
- Definition of measures for problem elimination
The implications for the product quality must be the main focus. The search for problem points must concentrate on this aspect. The points in the production process at which the product quality can be negatively influenced must be found.
|

5.1 Failure finding
Before failure finding is started, the production process (or the scope of the HACCP) should be described in sufficient detail. The description can be omitted if all those involved in the HACCP are very familiar with the system or the process. In any case, the system limits must be described.
Aspects for the description of a process or system:
- Composition (raw materials, additives, excipients, APIs)
- Nature and physico-chemical features (solid, liquid, pasty, emulsion, pH value, etc.)
- Processing steps
- Packaging (primary and secondary packaging)
- Storage conditions (temperature, humidity, light)
- Expiration dates
- Application
If the system is adequately described, careful and extensive failure finding of potential failures can be carried out. This is the first necessary condition for the success of a risk analysis, and for HACCP. With HACCP, all individual operations throughout the production process are to be checked to see if they can exert a negative influence on the product quality. Likewise, however, creative processes (brainstorming, brainwriting, etc.) can also be combined with HACCP to achieve the desired result.
For failure finding, it is crucial that all negative influences on the product quality are found. These can result both from the malfunctioning of machines, facilities or infrastructure or from human error. But the procedural instructions and operating procedures can also provoke mistakes. Every aspect of the process under investigation must withstand the questioning. There is no standardised catalogue of questions on which HACCP is based. Rather, each company must find its own way or approach to find the potential failures. The FDA has implemented a short catalogue of questions in the HACCP guideline in the food area, but unfortunately this does not help in the pharmaceutical industry as the requirements are too rudimentary. There is therefore currently no standardised catalogue of questions that accounts for the needs of the pharmaceutical industry.
In the established area of food processing, HACCP concentrates almost exclusively on hygiene. In the pharmaceutical area, this narrow constraint must be expanded, as there are significantly more risks in this area than just hygiene. For example, process management (temperatures, times, etc.), the initial weight of substances, the functionality of control systems and aspects such as preventative maintenance exert a great influence on product quality.
5.2 Evaluation of problem points
As shown in figure 19.B-24, the failure finding process (determination of feasible hazards) is followed by an evaluation of the problem points.
The aspects are evaluated using a two-tier classification
- Control point
- Critical control point
The following definitions are used for the control point and critical control point (source: HACCP Guidelines, FDA):
Control point (CP):
"Any point in a specific food system at which loss of control does not lead to an unacceptable health risk."
Critical control point (CCP):
"A point at which loss of control may result in an unacceptable risk."
In order to reach the evaluation of a control point or a critical control point, the following decision tree in figure 19.B-25 can be used.
|

With this grading, it is crucial that all processing steps be investigated systematically and consistently.
5.3 Definition of measures
If the decision has been made regarding which control points are present, then the measures to be initiated if control is lost must also be defined. This must usually be put into writing in operating procedures, so that if a loss of control does occur, it is clear what should be done. A loss of control occurs if the limits of a CCP are exceeded.
However, not only the measures, but also the verification procedure must be defined, in the form of test procedures, testing schedules or operating procedures (see figure 19.B-24). This step is crucial to be able to keep a process in the safe range during production. Here, it is important that the parameters with which a process can be brought back to the range that complies with the acceptance criteria, are known. Only if these control aspects are documented and communicated can the process be controlled, even if a critical process deviation occurs.
5.4 Documentation
As for every other risk analysis, HACCP too should be documented so that the results of the analysis are available at a later time. The following simple table (see figure 19.B-26) has proven useful for documentation purposes.
|

There are also more simple forms which only take into account the first three columns, for example, but this is less advisable as any further steps are not taken into account for CCPs and CPs and would have to be processed in a different form.
For documentation, the records of the discussions and above all of the decision must be as complete as possible. This is in the interest of anyone who has to represent the results at a later time (self-inspection, external inspection).
5.5 HACCP summary
CCPs are control points with which a process can be optimised. Whether or not sufficient control points have been defined is crucial for a process. If all significant control points are known and limits have been defined, the process is usually a controlled process. Process control is also an essential objective of HACCP. In order to achieve this objective, the process must be considered as a whole. HACCP offers a tool for localising CCPs and CPs through an established procedure in the field of hygiene.
Summary GMP risk analysis is a good instrument for establishing the GMP requirements in qualification and validation projects and for monitoring their realisation. With the consistent application of FMEA, HACCP or other forms of risk analysis, and the use of the results, the effort required for qualification/validation can be kept to the necessary level. The FMEA method, a company-specific risk analysis and the HACCP method are illustrated using examples, and their advantages and disadvantages are explained. It is especially important at the start of the project (procurement stage), to include a GMP risk analysis in the project sequence. |
