Risk Managemen Principles
|
The purpose of this chapter is to support the effective implementation of a risk management concept, and addresses the following questions:
|
The current climate in the pharmaceutical industry is influenced by the challenge of finding an appropriate balance between increased quality requirements, compliance with legal requirements, and cost pressure. The resources required for this are often very limited, which means that business processes must be organised more efficiently. If processes are reorganised at the expense of quality, the company may face damage to its reputation, for example, through product recalls. If savings are made in the area of compliance, recent examples clearly demonstrate that consequences of GMP breaches can incur costs in the region of hundreds of millions. The principle starting points for escaping from this dilemma lie in the targeted use of available resources and appropriate implementation of regulatory requirements. This begs the question: What is targeted and what is appropriate? This is where risk management can help. Terms such as Quality Risk Management (QRM), Risk Based Approach (RBA), Risk Analysis etc. have been familiar in the world of pharmaceuticals for some time. It should be made clear that these terms do not represent any fundamentally new developments. The new part of the concept is the actual approach of applying risk management systematically and across the board, and ensuring that the resulting advantages are fully utilised.
1 Objectives and advantages of risk management
The aim of risk management is to systematically evaluate processes and processing steps in terms of criticality, and subsequently develop appropriate measures to control and minimise risks. Corrective measures can subsequently be prioritised, their success becomes measurable, and the quality of products and processes is improved. This may mean that critical processes require more attention than previously, but for uncritical processes, the current workload can be justifiably reduced. Experience has shown that this concept not only incorporates quality and compliance, but also covers efficiency, environment, health and safety, as well as additional security aspects such as access control or data security mechanisms. In the following, however, emphasis is placed on quality and compliance. The advantages of risk management are listed below.
Advantages of risk management
- Applicable across the board
Risk management can be applied to all processes and products, and at all levels of a company. - Transparency
A consistent risk management process provides concrete statements about critical points and enables you to derive measures for minimising risk based on facts. - Integrated component of a QM system
Systematic communication of the critical process points to the QM system enables specific optimisation of the internal regulations. Elements of risk management are integrated into the quality assurance process. - Preventive rather than corrective: Action, not reaction
The systematic identification and evaluation of risk supports the prevention of prospective and retrospective activities (CAPA = Corrective Action Preventive Action). - Aggregation capability
The communication process between management and the authorities is encouraged (also see the FDA's risk based approach). - Risk awareness in staff behaviour
The introduction of a sustainable risk management concept requires that all employees involved are aware of the risks. - Integration of existing risk management approaches
It should be possible to integrate existing risk management approaches and activities into the overall system. - Standardised systematic approach to risk analysis
A range of application-case-specific standards for recording and evaluating risks supports the comparability of similar processes and the utilisation of synergies.
2 Regulatory environment
The current regulatory environment is influenced by two major initiatives:
- ICH Q9 Quality risk management (QRM), a global initiative providing a basis for the industry to evaluate processes and implement appropriate quality assurance measures.
- The FDA's Risk Based Approach (RBA), which primarily aims to optimise internal FDA procedures and inspection processes.
This chapter considers these two developments in more detail. The following describes the actual contents and their significance, whereby many parallels can be drawn between the two initiatives. Additional external specifications are subsequently listed and can serve as a useful reference for interested readers. These can provide a continuation or more detailed information.
- EN ISO 14971
Application of risk management to medical devices - FDA Guidance for Industry
PAT-A Framework for Innovative Pharmaceutical Development, Manufacturing and Quality Assurance (see chapter D.11 Guidance for Industry PAT -A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance) - FDA Guidance for Industry
Quality Systems Approach to Pharmaceutical cGMP Regulations
2.1 ICH Q9 Quality Risk Management
This document produced by the ICH (International Conference on Harmonisation) is currently (November 2005) in step 4 of the ICH approval process (see chapter E.8 ICH Q9: Quality Risk Management ). This is of great significance for the industry, since as an ICH document, it will be a worldwide standard and not only restricted to Europe or the USA.
 |
ICH Q9 does not contain any definitive new regulations. Instead it introduces a strategy, the basic principles, and a toolbox for evaluating processes in terms of risk, and standardising and documenting this evaluation. In contrast to other ICH documents, it is therefore more of a "How-to" document, which does not specify a "what", but instead contains suggestions on "how". As described in the introduction to ICH Q9, it serves as a "foundation or resource document that is independent of, yet supports, other ICH quality documents".
In this context, "processes" includes all processes - manufacturing as well as quality management processes. For the latter, Annex II of ICH Q9 lists concrete starting points that can be individually customised and further expanded. Examples are provided in chapter 10.E Fault tree analysis (FTA).
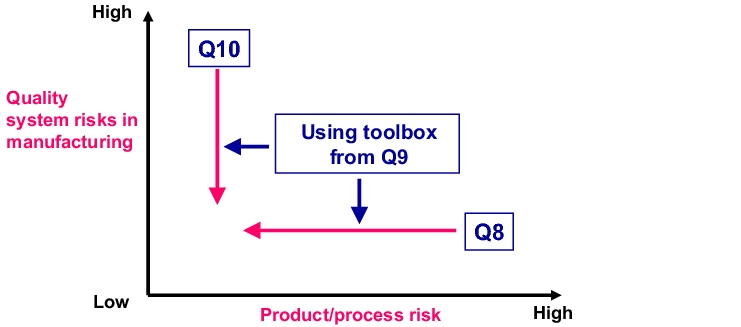
Of course, ICH Q9 is not the only current ICH document that covers this subject area. It offers more of a comprehensive approach, encompassing product development, manufacturing, and accompanying QM processes, which can be found in the three documents ICH Q8 Pharmaceutical Development, ICH Q9 Quality Risk Management and ICH Q10 Quality Systems for Continuous Improvement. The interaction between these three documents is shown in figure 10.A-1. While ICH Q8 clearly focuses on the products and requires the relevant product-specific measures during development (design space), ICH Q10 provides specifications for a (product-independent) quality management system.
2.2 The FDA risk-based approach
The FDA also has a clear aim of returning from an approach that has in part become highly formalised, to a more science-based outlook (Pharmaceutical cGMPs for the 21st Century: A Risk-Based Approach). This stems from the realisation that the current method inhibits innovation, and companies struggle to finance it in the long term. As a consequence, urgently required medicinal products may not become available on time. At the same time, the FDA itself also has limits in terms of capacity. For some time, it has not been unable to uphold the inspection intervals of two years as stipulated in specifications. This method also places emphasis on steering existing capacities towards the critical products, companies, methods, etc. This mainly affects cGMP inspections, where it can already be seen, for example, in the Quality Systems Approach (QSIT).
A good example of optimised cooperation between companies and authorities is provided by the FDA's considerations in the area of Change Control for manufacturing processes. Changes to complex products (e.g. proteins) manufactured in complicated processes are subject to a more intensive review by the authorities than previously. On the other hand, on a larger scale the FDA accepts a company's own change control system, if the quality system as such functions well, and the company can demonstrate a good understanding and monitoring of the relevant products and processes (see chapter 1.C.2 Change management system). As a result, well-monitored changes no longer need to be inspected or approved by the FDA. This enables capacities to be concentrated on critical products.
Further important and concrete results of this FDA initiative are described in the following.
21 CFR Part 11 - Scope & Application
In terms of the interpretation of Part 11 Requirements, this document puts an end to uncertainty and misunderstanding in the industry through the explicit statement that the work required for computer validation and working with electronic records should be adapted to a process-related (and hence more practical) quality measure. The requirements of 21 CFR Part 11 as such are not questioned. This is currently undergoing revision based on collected experience. Among other aspects, this affects:
- The procedure and scope of the validation itself: "We recommend that you base your approach on a justified and documented risk assessment and a determination of the potential of the system to affect product quality and safety, and record integrity."
- The definition of electronic records: "We recommend that, for each record required to be maintained under predicate rules, you determine in advance whether you plan to rely on the electronic record or paper record to perform regulated activities."
- The question of when and how an audit trail (= computer-generated log file) is necessary: "We recommend that you base your decision on whether to apply audit trails, or other appropriate measures, on a justified and documented risk assessment."
- The question of the most practical method of archiving: "We suggest that your decision on how to maintain records be based on predicate rule requirements and that you base your decision on a justified and documented risk assessment."
Process validation
In accordance with the Compliance Policy Guide (CPG) dated March 12, 2004, from the authorities' perspective, the validation of manufacturing processes will be considered as a more highly differentiated process in the future. Among other factors, this depends on when the validation has to be completed. The criteria mentioned include the company's history in terms of successful process validations, and a comparison with similar products or production processes. A company must be able to clearly demonstrate an understanding of the process resulting from the development, and that all necessary measures for process control have been met. If this is the case, marketing authorisation may be granted even before the completion of validation; although the first market batches still cannot be distributed until the validation has been successfully completed. In some cases, if the documented rationale is complete and in order, it may be possible to skip the production of conformity batches (with the agreement of the FDA).
On the other hand, however, the consequences are stricter if the process validation is considered insufficient, e.g. during an inspection. This not only results in a delay in the approval of the inspected product, but can also have a regulatory impact on other, similar products. This is particularly important for drug substances that are used in multiple products.
Pharmaceutical Manufacturing Research Project
With the support of Professors Dr Jackson A. Nickerson (Washington University in St. Louis) and Dr Jeffrey T. Macher (Georgetown University), the FDA initiated a project in 2001, of which the first sub-project involved the investigation of possible correlations between organisational, technical, and management-related conditions in the companies and cGMP compliance.
In the first step, the (voluntarily) participating companies were sent a detailed questionnaire, which recorded general company data (e.g. turnover, no. of employees, organisation), as well as information on the product range, handling of deviations, no. of batches/defective batches/complaints, number and outcome of inspections, number of submitted supplements, etc.
With the help of statistical evaluation methods, in the next step the manufacturing performance (quality, processing times, ...) is viewed in association with regulatory activities.
In return, each company receives an individual evaluation in the form of a scorecard, including a comparison with the average results of other comparable companies.
In a second sub-project, FDA data was classified and evaluated. Starting from warning letters, field alerts, recalls and similar events, it was possible to compile a profile of which product groups (e.g. parenterals, gel capsules, etc.) or which companies could be considered as critical in terms of probable non-compliance, and which therefore require more frequent inspection. In contrast, the inspection frequency for non-critical companies/products should be reduced.
Procedure for handling warning letters
For most companies, an FDA warning letter has very serious consequences, often including financial consequences. If the problem represents a real danger for patients, then the issuing of a warning letter is, of course, justified. However, this decision has previously rested with the field inspector (on-site inspector) alone, and the decision depends on many factors, including the overall course of the inspection. In a step to increase the objectivity of this process on a scientific basis, since March 2003, proposals for warning letters have been evaluated both by the Field Office (inspecting authorities) as well as the Center (regulatory authorities), before they are sent (for more information, see chapter B.3 Adress-Register).
Further objectives and aspects of the initiative
- Harmonisation of GMP requirements in 21 CFR Part 210 and 211 with EU, PIC/S
- Promotion of innovation: aseptic procedure, Process Analytical Technology (PAT), comparability studies
- Corrective and preventive action (CAPA), continuous improvement
2.3 Summary
It is rapidly becoming clear that the industry and the authorities share many common aims and interests which can best be overcome in a joint approach. The table below provides a summary of the most important common interests, as well as some of the differences
|
Quality risk management |
Risk based approach |
|
|---|---|---|
|
Motivation |
Increased requirements, cost and time pressure |
Increase in the number of companies requiring inspection without additional inspector capacity |
|
Aims |
Greatest benefit at the lowest costs with the use of limited resources |
|
|
Receive/create the opportunity to comply with own rules |
||
|
Improvement of quality and compliance |
Optimisation of inspection behaviour |
|
|
Methodology |
Structuring and prioritisation of risks |
|
|
Risk model |
Transparent, objective, systematic |
|
The area of quality risk management or risk based approach is a matter of great urgency and importance, both for the industry and for the authorities. The common goal is to provide patients with a sustainable, guaranteed supply of safe and effective medicinal products. In addition to the advantage of maximum transparency, the close cooperation between the authorities and industry enables a shared understanding and thus promotes effective implementation and mutual acceptance.
3 Science-based approach
The Science and Risk Based Approach initiated by the FDA has formulated the demand for a science-based and stable life cycle for pharmaceutical products. In accordance with the requirement to "know how it works", continuous quality should be guaranteed right from the development phase, and all critical aspects of the product and the manufacturing process should be analysed (Quality by Design, ICH Q8).
The introduction of the design space is designed to establish additional specification and parameter ranges within which limits the effect of the medicinal product can be safely achieved. This type of approach is ultimately intended to lead to greater regulatory flexibility and a sustainable reduction in the complexity of change management in favour of greater freedom in process and product optimisation. One aim of this approach is to achieve higher product quality with more flexible process design.
The introduction of PAT (Process Analytical Technologies) aims to introduce technologies that enable the variation of process parameters during the process, with the goal of maintaining consistent product quality.
The introduction of CAPA (Corrective Action/Preventive Action) (see chapter 1.C.4 Corrective and Preventive Actions (CAPA)) aims to implement systematic approaches that allow the derivation of measures for process and product improvement on the basis of retrospective and prospective analyses.
While the ICH Q8 Guide places emphasis on product quality, ICH Q10 is expected to contain specifications for the design of the quality system. It is already conceivable that a company is expected to have a quality system that not only requires the implementation of classical QM processes, but also in which measures are developed, for example, to describe the systematic handling of deviations identified in the QM processes. Furthermore, a company is also expected to have instruments in place for implementation and execution of the regulations.
3.1 Summary
ICH Q8, PAT, CAPA and ICH Q10: The application of a risk management concept is essential for the implementation of all these approaches. It forms the basis for targeted deployment of resources. Establishing a risk management process for all relevant stages of the life cycle, risk areas, and all company functions involved can offer a valuable contribution to high process quality and improved profitability.
|
Summary Quality risk management is increasingly expected by the authorities. It is no additional requirement, but as a measure it helps to correct fulfil of external regulations, efficiently and based on scientific facts. The authorities and industry are closely collaborating in order to jointly define the state of the art in this area. Important documents in this context are ICH Q9 Quality Risk Management, and the FDA's Risk-Based Approach. |
