Computer Validation. Regulatory aspects
|
Here you will find answers to the following questions:
|
The most important regulatory requirements are:
- The EU-GMP Guideline, Annex 11
(see chapter C.6.11 Annex 11 Computerised Systems) - 21 CFR (Code of Federal Regulations) Part 11
(see chapter D.1 21 CFR Part 11 Electronic records; electronic signatures) - PIC/S document PI-011
(see chapter F.3 PIC/S Guidance Good Practices for Computerised Systems in Regulated "GXP" Environments (PIC/S PI 011))
9.B.1 Europe
Several parts of the EU-GMP Guideline (see chapter C.4 Part I Basic Requirements for Medicinal Products) deal with computerised systems. In particular, it is worth mentioning chapter 4.9, which permits the use of computerised systems in data recording, and chapter 5.29, which states that in completely computerised storage systems, the labelling does not absolutely have to be included in a legible format on the label.
|
Annex 11 of the EU-GMP Guideline |
|---|
|
0. Principles |
|
1. Personnel |
|
2. Validation |
|
3. System and environment |
|
4. System description |
|
5. Software |
|
6. Automatic accuracy check |
|
7. Validation before start-up, parallel operation |
|
8. Authorisation for entering or changing data |
|
9. Entry of critical data |
|
10. Identity of the operator when entering critical data, audit trail including reason for the change |
|
11. Alterations to a system |
|
12. Printed copies |
|
13. Securing of data against wilful or accidental damage |
|
14. Backup copies at a separate and secure location |
|
15. Procedure in the event of a breakdown |
|
16. System failure and breakdown |
|
17. Analysis and correction of errors |
|
18. Outsourcing |
|
19. Computerised release for sale or supply of medicinal products |
However, the most important guidelines are the 20 paragraphs of Annex 11 of the EU-GMP Guideline (see chapter C.6.11 Annex 11 Computerised Systems and figure 9.B-1). In 1996, the IT focus group of the APV (International Association for Pharmaceutical Technology) published an interpretation of Annex 11, which was included in the GAMP Guide.
This Annex emphasises that the use of computerised systems does not change the necessity of complying with good manufacturing practice. If a computerised system replaces a manual operation, this must affect neither the product quality nor quality assurance. Inclusion of the user is very important, since if user involvement is limited, certain aspects of the earlier system may be lost. When replacing manual systems with automated systems, it is suggested to run both systems in parallel for a certain period of time.
Key personnel should work closely together with IT staff, and should be suitably educated in terms of planning and use of the system. The necessary subject knowledge for the validation, installation, and operation of computerised systems should be available.
The scope of the required validation partly depends on the application area of the system. Validation and testing should be performed before the system is used. This should include a detailed description of the system and should always reflect the latest state of the art. The life cycle phases, mentioned in Annex 11, planning, specification, programming, testing, start-up, documentation, operation, monitoring, and change management are named.
The requirements for the environment of a computerised system are similar as for all other equipment: A suitable environment in which neither humidity, dust, nor heat are permitted to negatively influence the system.
Since, in accordance with Annex 11, software is identified as a critical component of a computerised system, it should be created in conformance with a quality assurance system. This should then be verified during audits (see chapter C.6.11 Annex 11 Computerised Systems).
A plausibility check should test the accuracy of the input and processing of data. Manual entries of critical data (e.g. weight and batch number of an API for dispensing) should also be tested separately for accuracy. This can be performed by a second operator or a validated electronic method, e.g. bar code reading.
Any entry or alteration of data, e.g. batch release for placing on the market (Annex 11, point 19), should only be performed by authorised persons. Suggested measures to protect against unauthorised data entry are the use of locked cupboards (such as those often used in programmable logic controls), ID cards, or personal codes. With these methods - as explained in point 10 of Annex 11 - the system must check the identity of the operator entering or confirming the data. Any alteration of data should be authorised and recorded together with the reason for the change. Annex 11 requires an audit trail, which is a complete record of all entries and amendments to data. The audit trail is nothing more than the fulfilment of the requirement for traceability, as demanded by the principles of chapter 4 of the EU-GMP Guideline. (See chapter C.4 Part I Basic Requirements for Medicinal Products.)
The issuing, cancellation and changing of authorisations for entering and amending data, including changes to personal codes, should be established in writing. Access attempts by unauthorised persons should be documented, or access should be blocked after a certain number of invalid attempts.
The change management in point 11 of Annex 11 determines that alterations to a computerised system must be made in accordance with a defined procedure. This procedure must include specifications for authorisation of the relevant persons, validation, checking, approval, and implementation of the change. Errors should be handled in the same way as changes, but must also be subject to further investigation.
Point 13 of Annex 11 makes reference to item 4.9 of the EU-GMP Guideline and requires that the system must be capable of enabling meaningful printouts of the electronically stored data. Furthermore, the data must be physically or electronically secured against wilful or accidental damage and must be checked for accessibility, durability and accuracy. This also includes the creation of regular backup copies, which should be stored at a separate and secure location for as long as necessary.
Suitable alternative arrangements must be in place in the event of a system breakdown. This affects all business-critical processes as well as the compilation of recall lists. These alternatives should be defined and validated.
When outsourcing activities, formal agreements must be drawn up which clearly define the responsibilities of the external company (see chapter 9.G.2 Service level agreement).
9.B.2 PIC/S
The PIC/S recommendation PI 011 "Good Practices for Computerised Systems in Regulated 'GxP' Environments" (see chapter F.3 PIC/S Guidance Good Practices for Computerised Systems in Regulated "GXP" Environments (PIC/S PI 011) essentially contains the same points as Annex 11 of the EU-GMP Guideline and 21 CFR Article 11. Chapter 24 provides detailed help for inspectors (the target audience of this guideline) on how to inspect computerised systems in the GxP environment.
9.B.3 USA
In the US GMP regulation 21 CFR 211, § 68a) on "Automatic, mechanical and electronic equipment", states that the use of this equipment is permitted as long as it is regularly calibrated, inspected, or checked (see chapter D.1 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceuticals).
§ 211.68b requires change control for the entered data and corresponding control of authorisations. Furthermore, the data must also be checked for accuracy, although these tests depend on system complexity and reliability. In this context it is notable that the criticality of the data is insignificant. The compilation and retention of backup files are described here, although certain data, such as calculations performed during laboratory analysis or other intermediate results that are automatically deleted, are not subject to this backup.
It is sufficient to retain records, which are to be inspected directly at the establishment where the described activity took place, in a legible format on computers in that location (subpart J "Records and Reports", § 211.180 paragraph a). More detailed requirements specific to electronic documents and electronic signatures are contained in 21 CFR Part 11 (see chapter D.1 21 CFR Part 11 Electronic records; electronic signatures). There is considerable overlap between these requirements and those of Annex 11 of the EU-GMP Guideline.
In addition to the regulations, in the USA there is also a large number of guidelines that are considered standard for computer validation, e.g. the Guide to Inspection of Computerized Systems in Drug Processing (see chapter D.7 Guide to Inspection of Computerized Systems in Drug Processing). One of the most important guidelines with reference to the validation of computerised system is the Guidance for Industry: Electronic Records and Electronic Signatures - Scope and Application (see chapter D.12 Guidance for Industry Part 11, Electronic Records; Electronic Signatures - Scope and Application). This publication places a relative perspective on the requirements of 21 CFR Part 11 and places a risk-based procedure at the forefront. This guidance states that the FDA is planning to revise 21 CFR Part 11 and may consider a change to the legislation. In the meantime, the FDA will interpret 21 CFR Part 11 very narrowly.
In particular, the requirements in terms of
- Validation,
- Audit trail,
- Retention of documents,
- Copies of documents,
- Use of legacy systems
should be implemented only if there is acertain risk.
9.B.4 Electronic signature and electronic records
The following table (figure 9.B-2) compares the official regulations of Europe, the USA and the PIC/S in terms of electronic records/electronic signatures (21 CFR Part 11).
|
21 CFR Part 11 |
EU-GMP Guideline |
PIC/S |
|---|---|---|
|
11.10 e) Availability for inspection |
Mentioned implicitly |
5.3 |
|
11.3.b.5 Digital signature |
Not mentioned |
21.10 |
|
11.3.b.8 Handwritten signature |
Not mentioned |
Referenced 21.5; Glossary |
|
11.3.b.7 Electronic signature |
Not mentioned |
21.6; 21.7; |
|
11.3.b.6. Electronic records |
4.9 |
Referenced to the FDA definition |
|
11.3.b.4. Closed system |
Not mentioned |
Not mentioned |
|
11.3.b.9. Open system |
Not mentioned |
21.5 |
|
11.10.a Validation of systems |
4.9, Annex 11.1, 2, 7 |
23.6; 23.8; 23.10 |
|
11.10.a Identification of changes |
Not mentioned |
Not mentioned |
|
11.10.b Creation of copies |
Annex 11.12 |
21.13 |
|
11.10.c Protection |
4.9 |
21.1 |
|
11.10.d Limiting system access |
4.9, Annex 11.8 |
21.13 |
|
11.10.e Audit trail |
Annex 11.10 |
21.13 |
|
11.10.f Compliance with step sequences |
Not mentioned |
Not mentioned |
|
11.10.g Authority checks |
Mentioned implicitly |
Not mentioned |
|
11.10.h Input device checks |
Not mentioned |
Not mentioned |
|
11.10.i Training and experience |
2.8, 2.9 |
15.3, 21.10; 22.2, 22.5; 22.6; 22.7 |
|
11.10.j Signature accountability |
Not mentioned |
12.1; 21.10 |
|
11.10.k.1 System documentation |
4.2; 4.5 |
Not mentioned |
|
11.30 Encryption for open systems |
Not mentioned |
19.4; 21.11 |
|
11.50 Signature manifestations |
Not mentioned |
21.10 |
|
11.70 Signature/record linking |
Not mentioned |
Mentioned implicitly |
|
11.100.a Uniqueness of signature |
Not mentioned |
21.9 |
|
11.100.b Identity verification |
Not mentioned |
Not mentioned |
|
11.100.c Certification to the FDA |
Not mentioned |
Not mentioned |
|
11.200.a.1.i 2 distinct components |
Not mentioned |
21.9 |
|
11.200.a.1.ii Series signing |
Not mentioned |
Not mentioned |
|
11.200.a.2 Non-transferability |
Not mentioned |
21.9 |
|
11.300 Administration of signatures |
Annex 11.8 |
19.3 |
|
11.300.a Maintaining uniqueness |
Not mentioned |
21.9 |
|
11.300.b Password ageing |
Not mentioned |
19.3 |
|
11.300.c Loss management |
Annex 11.8 |
19.3 |
|
11.300.d Transaction safeguards |
Not mentioned |
Not mentioned |
|
11.300.e Testing of signature devices |
Not mentioned |
Not mentioned |
|
Not mentioned |
Critical data 4.9, |
Critical data 20.2 |
The requirements of 21 CFR Part 11 overlap to a great extent with the requirements of the EU and PIC/S. It is notable that specific handling of critical data does not deserve a mention in the FDA documentation, while more technical details such as signature ageing, regular inspection of input devices, and transaction protection are not mentioned in the PIC/S.
9.B.5 GAMP® Good Automated Manufacturing Practice
The GAMP® Guideline is not an official requirement, however, the authorities work together with industry representatives and experts from the computer industry in various committees on this "quasi" standard . In industry practice, the GAMP Guide is consulted to very frequently.
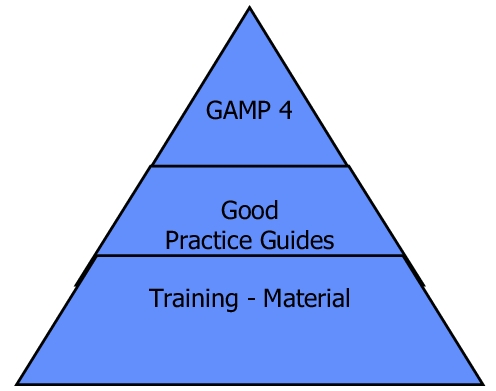
GAMP® consists of a GAMP® Guide, which is currently available in version 4 (source: www.ispe.org). In addition, various SIGs (Special Interest Groups) are refining the individual chapters and are publishing them with examples intended as Good Practice Guides. These are distributed and presented along with training material at ISPE conferences. figure 9.B-3 shows the basic structure of the GAMP publications.
 |
|
Summary: The strictest requirements for the validation of computerised systems are formulated by the 21 CFR Part 11 and enforced by the FDA. In Europe, these are based on Annex 11 of the EU GMP Guideline. The PIC/S (Pharmaceutical Inspection Cooperation Scheme) and GAMP are not authorities, but are committees that develop guidelines for industry and authorities. |
ISPE = International Society of Pharmaceutical Engineers (www.ispe.org) .
