8.H Documentation
|
Here you will find answers to the following questions:
|
According to section 7.4 of the PIC/S guideline (PIC/S PI006) entitled Documentation, a validation protocol and a validation report must be compiled. Further, the cleaning process must be described in an operating procedure and also documented each time it is carried out. The formal and content aspects of the documents to be compiled will be dealt with in more detail below.
8.H.1 Validation protocol
Section 7.4.1 of the PIC/S guideline (PIC/S PI006) contains a very detailed list of information that should appear in the validation protocol.
All the information required here must be included in the cleaning validation documentation. However, the practical application of this list for the individual validation protocols must be critically assessed - as many of the items listed may be defined in a master document or may be described in other documents, which means that their repetition in each individual validation protocol would result in unnecessary duplication.
The required contents of the validation protocol according to the aforementioned PIC/S guideline are listed in figure 8.H-1 and commented on below.
|
Contents of the validation protocol: |
|---|
|
If a master plan is compiled for the cleaning validation project that includes all aspects mentioned in chapter 8.C Cleaning validation master plan, definitions and provisions that generally apply for the cleaning validation can be dealt with once in this plan and must not be repeated in every validation protocol.
Examples: the definition of the objective, responsibilities for the implementation of the validation and the compilation and authorisation of documents, as well as revalidation specifications.
Decisions that take all products and equipment into consideration, and are therefore only clear and comprehensible within the context of all available data, should also be laid down once in the master plan as a higher-level document. Examples: the application of bracketing through selection of critical substances; the choice of the sampling procedure based on the substance characteristics and the construction of the production equipment, together with the rationale for the choice of acceptance criteria based on comparative limit calculations.
The calculation of limits for the acceptable residue in the subsequent product, as well as in the sample, is most efficiently carried out once in the master plan (e.g. in an appendix) rather than for each individual validation protocol. The critical product "A" described in chapter 8.D Establishing the scope of validation which ist manufactured using nine different types of production equipment (see figure 8.D-5) may be used to clarify this point. Following the equipment-related approach for cleaning validation, nine validation protocols would need to be compiled, each containing the calculations shown in chapter 8.E Acceptance criteria and limit calculation and figure 8.E-3 to figure 8.E-5.
In contrast to the calculation itself, the calculated limits in the subsequent product as well as in the individual sample should be described in the validation protocol for each critical product manufactured on the relevant production equipment.
Data related to the validation of analytical methods should primarily be included in the relevant validation reports or in the analytical procedures. Data for the limit of detection and limit of quantitation as well as recovery are examples of this. In the validation protocol, a reference to the analytical procedures, according to which the residues of the relevant critical substance must be determined is sufficient.
Data related to the cleaning itself should be part of the cleaning instructions. The number of cleaning operations that must be consecutively carried out, and also the holding time between the end of production and start of cleaning. In this context it is important to point out that the maximum holding time specified in the cleaning instruction must be taken into account when carrying out the validation. All of the cleaning instructions on which the validation is based must be specified in the validation protocol. In the most straightforward case this would be a single cleaning instruction (e.g. sieving machine or container), but several cleaning instructions may also be necessary for individual accessories that are validated jointly as one functional unit (e.g. tablet press and deduster). Furthermore, different cleaning instructions for various products may be described in one cleaning instruction (e.g. same process sequence, different cleansing agents). It is important here to ensure that the validation takes all the procedures described into account.
A precise description of the production equipment to which the cleaning validation refers must be included in each validation protocol. To make sure the equipment is clearly identified, information such as the inventory number and location can be provided in addition to the unit designation and type. It is important to specify all production facilities and process equipment that apply for the respective cleaning instruction and therefore also the validation.
The definition of sampling locations is a very important part of the validation protocol. Experience has shown that a clear and unambiguous designation for an accessory or a critical location is not easy, as staff frequently use different names to describe the same items. Therefore, drawings of units or photos showing the precise sampling locations are valuable additions to the sampling plan.
It is advisable to lay out the sampling plan as a form in which the sampling is documented and the analytical results of the individual samples noted. This provides a compact and clear summary of all important information obtained during the individual tests. Figure 8.H-2. shows what this form might look like.
 |
The validation protocol should be approved by quality assurance and management. This ensures compliance with official requirements and also ensures that the necessary resources have been noted and will also be made available.
8.H.2 Validation report
Once the validation is complete, a final validation report which refers to one specific validation protocol must be compiled. The report should summarise the results and particularities of the relevant validation study, and, as a conclusion, provide an assessment as to whether the investigated cleaning procedure was successfully validated. As is the case with the validation protocol, the validation report must be approved by quality assurance and management.
In order to present a summary of the validation study results and to assess them, the sampling plan must be referred to. This means that the sampling plan is a component of both the validation protocol (blank form as guideline for sampling) and the validation report (completed form that documents the sampling and sampling results). If the protocol and report are compiled as separate documents, it will be necessary to write these in duplicate as the information will overlap.
Apart from the sampling plan, the validation protocol includes many additional specifications that must be referred to in the validation report, again making repetition unavoidable
|
Documentation of a cleaning validation study |
|---|
|
Cover sheet with circulation for approval 1. Circulation for approval: validation protocol (sections I, II, IV) 2. Circulation for approval: validation report (sections I - V) |
|
I Basic data for validation I.1 Production equipment
I.2 Cleaning instructions
I.3 Scope of validation
I.4 Sampling methods
I.5 Analytical methods
I.6 Acceptance criteria
|
|
II Sampling plans II.1 Visual check
II.2 Test for active pharmaceutical ingredient residues
II.3 Test for cleansing agent residues
II.4 Test for microbiological contamination
|
|
III Results and evaluation III.1 Results:
III.2 Distinctive features and measures III.3 Summary |
|
IV Appendix to validation protocol
|
|
IV Appendix to validation report
|
.
To arrange the validation documentation clearly and efficiently, the compilation of a single document incorporating both protocol and report is recommended. The following suggestion shows how this kind of document might be structured in terms of form and content (see figure 8.H-3).
Section I entitled Basic data for validation contains all information and provisions relevant to the validation study.
The sampling plans may be found in section II (initially blank forms). According to section 7.3.6 of the PIC/S guideline (PIC/S PI006), evidence of validation must be provided by at least three consecutive cleaning cycles with positive results. It follows that in each case, three forms for visual control tests for active pharmaceutical ingredient residues, and tests for cleansing agent residues and microbiological contamination must be regarded as the minimum scope of this section. If several critically-graded products are manufactured using the production equipment concerned, or if several cleansing agents are used, the number of tests and forms required increase accordingly. Product and equipment-specific information such as the type and quantity of solvent to be used for the swab or rinse test as well as distinctive features of the test implementation (disassembly of facility parts, aids to be used, or similar) can be included under Information on sampling.
Drawings of equipment and/or photos in which the sampling locations are clearly identified can be found in section IV.
Sections I, II and IV are approved as a validation protocol by an initial circulation for approval.
Once the validation is complete sections III and V must be compiled. In the interests of clarity, section III should be kept concise and should only contain information that is relevant for the validation assessment. A detailed and comprehensive presentation of the individual sampling results in tabular form may be appended in section V.
If distinctive features or deviations happened during the validation, all related activities and resulting measures must also be described in section III. Related documents must be appended in Section V.
A brief summary that assesses the overall result of the validation should be included at the end of section III.
Once the validation is complete, the entire document incorporating sections I - V is then submitted for approval as a validation report.
8.H.3 Other documents
According to the PIC/S guideline (PIC/S PI006), a cleaning instruction and a cleaning report must be compiled in addition to the validation protocol and validation report.
The contents of the cleaning instruction are not specified in the aforementioned PIC guideline (also see chapter 8.B.2 Compilation of cleaning instructions).
However, it is specified what kind of information on each cleaning operation should be documented:
- cleaned area or cleaned production equipment
- person carrying out
- date of cleaning
- cleaning instruction
- last product manufactured prior to cleaning
One means of implementing this requirement in practice is to produce a cleaning label to be attached to the cleaned equipment after completion of the cleaning. To ensure traceability, this label may be appended to the manufacturing documentation of the subsequent product.
Additional documents in the context of cleaning validation are not required according to the PIC/S guideline (PIC/S PI006).
On the other hand, according to the FDA inspection guideline "Guide to Inspections of Validation of Cleaning Processes", "written general procedures on how cleaning processes will be validated" are required in addition to the validation protocols and reports.
This requirement can be satisfied by compiling the aforementioned master SOP cleaning validation. This master SOP should address all important aspects of the cleaning validation but the emphasis here is more on fundamental provisions rather than specific details of implementation. This must be reserved for the cleaning validation master plan or the individual validation protocols, if a plan of this kind is not compiled. Figure 8.H-4 provides a proposal for structuring the contents of a master SOP cleaning validation.
|
Master SOP cleaning validation (proposed structure) |
|---|
|
1. Definition of objective 2. Responsibilities 3. Area of application 4. Bracketing
5. Acceptance criteria
6. Sampling procedures
7. Analytical methods
|
|
8. Integration of cleaning validation in existing quality assurance systems qualification (equipment, systems)
|
In addition to these documents required by the authorities, the compilation of operating instructions for the individual sampling procedures is recommended as an aid to practical implementation. The analytical methods for determining residues also belong to the documents that must be compiled in the context of cleaning validation.
Finally, the cleaning validation must be appropriately integrated into existing quality systems, e.g. deviation system, change control and revalidation. In this case it is recommended that a separate section about cleaning validation should be provided in the respective master SOPs.
All documents that must be compiled in the context of cleaning validation are presented once more in figure 8.H-5.
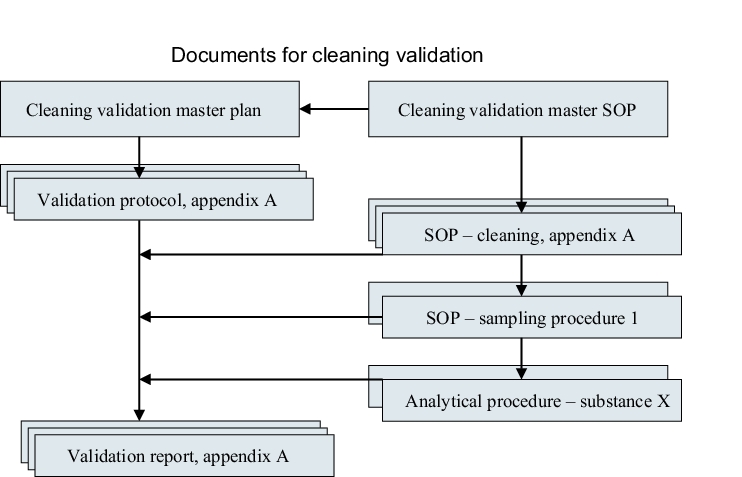 |
|
Summary |
|---|
|
The following documents should be compiled in the context of cleaning validation (see figure 8.H-5):
|
|
The validation protocol should contain the following product and equipment-specific data (see figure 8.H-3):
|
|
The validation report should contain the following information (see figure 8.H-3):
|
|
In order to avoid unnecessary repetition in the validation protocols, general provisions for cleaning validation should be made in higher-level documents (master SOP or master plan). Substance specific analytics ans analytical methods validation should be contained in the relevant analysis instructions and validation reports. General sampling specifications should be contained in operating instructions for the relevant procedure. References to the relevant documents are provided in the validation protocol. To avoid unnecessary repetition in the validation reports and to limit the number of documents that must be compiled to a manageable level, the validation protocol and report may be combined in one document containing all necessary information. |
