|
Here you will find answers to the following questions:
|
1 Responsibilities and task assignment
From a legal point of view, performance of process validation lies within the responsibility of the "Qualified Person" for EU countries (for other countries, please refer to the respective local regulatory guidelines). In any case, the responsible person for process validation must be specified in the individual validation master plan (VMP) of a company (see chapter 7.F Validation master plan). In practise however, this person will rarely be able to carry out the complex planning, implementation and documentation tasks himself. These tasks may, for example, be handed over to a validation manager who coordinates the overall validation project and maintains an overview of the many sub-tasks. The validation manager may appoint a validation team made up of representatives from the specialised departments who are entrusted with the practical work, e.g. production, engineering, technical, process engineering, documentation and quality control units. The GMP approach to this form of delegation always assumes that the responsible person delegates the task to competent staff and also subsequently ensures that the task assigned is progressing. Tasks and responsibilities delegated must be documented in writing, e.g. in the validation master plan or validation protocol, validation policy or relevant SOPs. The tasks typically performed by the validation manager are listed in figure 1.
|
Typical tasks of a validation manager |
|---|
|
It is not difficult to imagine that in the case of large validation projects, e.g. starting up new manufacturing sites, these tasks are too wide-ranging to be dealt with on top of day-to-day business tasks. The creation of full-time positions for persons responsible for validation has therefore proven to be an effective approach. Smaller recurring validation projects (e.g. revalidation) often tend to run more smoothly as well when dealt with routinely by validation managers rather than if they were added to existing high management workloads.
All recent rules and regulations for qualification and validation emphasise the importance of careful, documented planning of validation activities. However, the subject of the validation dictates how the planning and implementation of the validation is organised. Does the case in hand concern the handover of a validated procedure with a low level of technical know-how required and a "low-risk" product (see chapter 7.G Risk analysis) to another production site, or is it the validation of a new operation that deals with innovative, technologically highly-complex dosage forms? The organisation of the specific pharmaceutical company must also be taken into account. Ultimately, the additional structures required to carry out the validation should support normal business activities and interfere with these as little as possible as otherwise quality would be affected.
This is also the reason why no recommendable one-fits-all "standard validation protocols" exist: each company is organised differently and each validation project is different to the next. If the manufacturing procedures for two or more products of one manufacturer are in fact very similar, a joint validation may be undertaken according to a "matrix concept" (see chapter 7.F Validation master plan). In all other cases a universally applicable "standard validation concept" - frequently requested by persons not so experienced in process validation - would carry a high risk: for many products, to work through a standard plan would result in unnecessary costs and workload without adding any benefit to the quality of the final product. Unlike for other products, sticking to the same standard plan might be insufficient - e.g. for highly-innovative products with complex manufacturing procedures possibly containing active pharmaceutical ingredients with a narrow therapeutic range. Legal responsibility for the extent of validation activities is therefore placed firmly in the hands of pharmaceutical companies - they should know their manufacturing processes and be able to demonstrate through documentation that they are well controlled.
It is particularly important to clearly define the areas of responsibility if validation tasks are outsourced (partially or fully) to contract acceptors (see chapter 17.A Contract manufacture).
|
Arrangements that must be made when assigning validation activities to a third party |
|---|
|
In this case, a number of different scenarios are possible:
- A pharmaceutical manufacturer that has its own manufacturing authorisation outsources the validation of a manufacturing process (or parts thereof) to an external company, e.g. a consultant. In this case, the contract giver remains responsible for the validation.
- A pharmaceutical manufacturer that has its own manufacturing authorisation hands a manufacturing process and its validation over to a contract manufacturer. In this case, a contractual agreement defines whether the contract giver or the contract manufacturer is responsible for the validation.
- A pharmaceutical company (holder of marketing authorisation) without its own manufacturing authorisation outsources the validation of a manufacturing process to a contract manufacturer. The responsibility for this validation lies with the contract manufacturer.
Where validation tasks are assigned to third parties, the validation manager must rise to the additional challenge of ensuring effective moderation, clear agreements and a fast flow of information between the contract giver and the contract acceptor (see figure 2). More often than not, the information and experience at the disposal of both parties is different and their philosophy and terminology does not correspond. It is therefore important that both contractual partners have defined their respective intentions (in the validation master plan) and also agree contractually about what they expect from one another. To do this, a detailed agreement must be drawn up in writing that defines the scope of the service to be provided and the general conditions (e.g. GMP, DIN/ISO 9000ff, see chapter 1 Quality Management) the contract acceptor must fulfil.
2 Validation team
Validation projects are usually planned and executed by a validation team led by a validation manager (see figure 3). The team can also be assisted by an external consultant if necessary. However, decisions must ultimately be made by the team or by representatives of the departments involved. The following departments/functions should be represented in this validation team to make the aforementioned contributions: (This should be regarded as an example, as the responsibilities in the departments vary greatly depending on the size of the company and the manner in which it is organised):
|
Structure of the validation team (example): |
|---|
|
- Technical department, engineering, EDP department
Maintenance status of facilities and machines
Status of qualifications
Technical documentation
Computer validation status - Documentation department, regulatory affairs department
Current master production record (conforming with application file for marketing authorisation)
Current test procedure
Current validation master plan
Special regulatory and (if relevant) country-specific requirements - Development department, process technology
Planning of development and pilot batches (if relevant)
Results of development and pilot batches - Head of manufacturing/production
Responsible for implementation of validation
Provision of resources
Approval of validation protocol and validation report
Approval of master production record for routine manufacturing
Manufacturing of validation batches, additional sampling according to sampling plan - Production planning
Planning in of sufficiently large time windows for use of facilities and machines - Head of Quality Control
Validation status of analytical methods
Approval of analytical methods
Capacity planning for analytical tests - Control laboratory
Implementation of analytical work - Microbiology
Status of environmental monitoring
Assessment of microbial reduction during cleaning validation
Microbiological assessment of holding times - Quality assurance
Approval of validation protocol and validation report
Approval of product specifications
Approval of validation batches for clinical studies, for commercial use or as registration/application samples (if relevant)
Control of compliance with internal and regulatory guidelines, e.g. through internal audit (control of equipment documentation, logs, SOPs, sanitising, compliance with gowning procedures,...)
Approval of master production record for routine manufacturing
Change control
3 Timing of validation
In general, only pharmaceutical products manufactured according to validated manufacturing processes may be distributed. This means that by the time the product is launched, the process validation must be complete. The time between the submission of the file for marketing authorisation and the issue of approval is very frequently used for process validation activities. The EMEA "Note for Guidance on Process Validation" provides more detailed information on the validation data for specific products that must be included in the application file at this stage:
For specific medicinal products manufactured according to standard procedures, it is sufficient if data from a pilot scale validation and the schedule for the commercial scale validation are available at the time of submission.
On the other hand, for "non-standard products", such as medicines with modified release, specific lyophilisates, micro capsules or aseptically manufactured products, it cannot necessarily be assumed, that processes are easily transferable from pilot scale to production conditions. It is therefore expected that at least 1 or 2 batches will have been subjected to a production-scale validation before the files for marketing authorisation are submitted. This data must also be supported by pilot batches and a range of products that have been consistently manufactured under similar process conditions. For non-standard sterilisation methods and aseptic manufacturing methods, the data from three consecutive production-scale batches must be submitted as a prerequisite for approval (EMEA "Note for Guidance on Process Validation", paragraph 4.4).
4 Prerequisites for carrying out a validation project
The process validation is not an isolated action in an otherwise unaffected environment. Instead it has far-reaching consequences that have already been explained in more detail at "Revalidation" (see chapter 7.D Revalidation). Therefore several important prerequisites must be met in order to ensure proper implementation of the process validation concept.
|
Prerequisites for carrying out a process validation |
|---|
|
This includes:
- An approved validation master plan (see chapter 7.F Validation master plan)
- A fully developed product and developed, optimised process:
In this context, the pressure to achieve shorter development times due to international competition unfortunately may have fatal consequences: shortcuts taken in the early development stages that (apparently) save time mean that badly developed processes are carried through to the production handover stage. Attempts are subsequently made during validation phase to make improvements that should definitely have been carried out at an earlier stage: in addition to process optimisation, the tasks of specification of critical process parameters, specification of ranges, of determination of margins of error and "challenging" are tried to be carried out simultaneously. The problems that emerge have a considerable impact on material and time costs. In addition, these kind of "last-minute experiments" tend not to instil confidence in the authorities that the process is well understood and adequately controlled. Finally, it must be remembered that these kinds of "validation batches" cannot simply be made to "disappear" if they do not comply with the specifications. These cases involve a large amount of expenditure in finding, eliminating and documenting errors and must be documented as OOS batches in the manufacturing history. The three final validation batches must demonstrate unequivocally that the manufacturing procedure is reproducible.
Likewise the latest recommendations on qualification and validation (EMEA "Note for Guidance on Process Validation", paragraph 3 and ZLG Aide Mйmoire entitled "Inspection of qualification and validation in pharmaceutical manufacturing and quality control", paragraphs 6.1 and 6.3) point out that critical steps and critical process parameters, challenge tests, proof of suitability (feasibility) and robustness of the process must be established at the development stage. They are in fact prerequisites for (and not part and parcel of) the process validation. - Sufficient information from development stage to clearly define critical process parameters and normal operating ranges:
The validation should verify the normal operating parameters (cf. EMEA Note for Guidance: "within their specified design parameters") under realistic conditions but should not test the process to its limits ("challenge"). If "challenge tests are planned within the scope of the validation, these should not test the process, to its limits. Instead, effectiveness of control measures, performed in order to detect, eliminate and/or prevent product failures is to be shown (cf. ZLG Aide Mйmoire entitled "Inspection of qualification and validation in pharmaceutical manufacturing and quality control", paragraph 6.3.1). Relevant examples are function tests for detection systems such as automatic cameras used to control level and presence, or similar systems. Strictly speaking, the purpose of these "challenge tests" - or function tests - is to qualify operating equipment rather than validate processes.
By the same token, the term worst case is used in conjunction with process validation. The worst case is defined as "a condition or a set of conditions encompassing upper and lower processing limits and circumstances, within standard operating procedures, which pose the greatest chance of product or process failure when compared to ideal conditions. Such conditions do not necessarily induce product or process failure" (see PIC/S PI006). In other words: process validation is expected to cover normal variability under real life conditions, but it is not necessary to build in "artificial events" or extra risks in validation runs. Or in the words of the ZLG Aide Mйmoire "Inspection of ....quality control": validation is to be carried out under conditions "close to reality". For example, raw materials from the various suppliers (approved for the specific product) should be used in the three validation batches, which should be produced and/or packaged by different personnel (e.g. different shifts). The purpose of this approach is to prevent everything running "perfectly" during the three validation cycles, where everything is planned with the utmost care in advance, the most experienced people on hand, no holding times (as the job needs to be finished quickly), but disregarding production-related variations which will occur later in "real life".
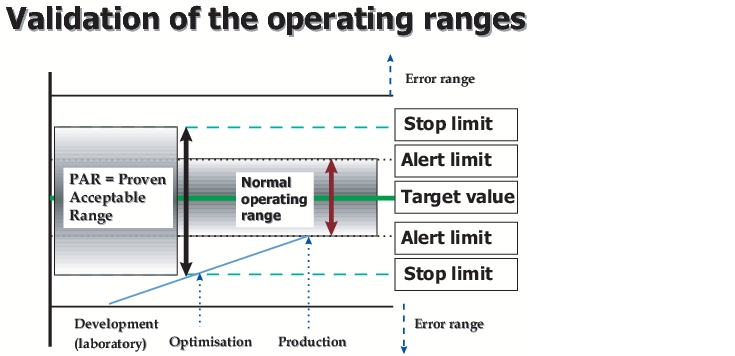
The operating parameters must have been established during the course of development as well as the PAR (Proven Acceptable Range - range in which the process parameters may demonstrably fluctuate without affecting the quality of the final product). In practise, however, the situation with respect to equipment during development is sometimes so different from the reality of production that development data are not directly transferable. In these cases, the data required must be obtained before the validation is carried out from pilot batches (production equipment, but with smaller batches).
In figure 5, a graphic representation of the correlation between the normal operating range and the PAR range is shown. The "normal operating range" is the range that must be validated according to law. This range is also stated in the application file. For day-to-day operations however, additional stricter "internal operating ranges" and/or "internal specifications" are often used. This is to ensure that in case of a deviation from the "operating range", this is still not outside the validated = approved range (i.e. the product can still be put on the market).Figure 5 Validation of the operating ranges
- Qualification of facilities and machines used as well as calibration of the measuring equipment (see chapter 6 Qualification):
If facilities and equipment are not qualified and measuring equipment is not regularly calibrated, the assessment of machine settings is not reliable at all. If, at a later date, it becomes apparent that the values read out from the displays do not in fact correspond with the actual values, this would render the results of the validation invalid and require revalidation.
The significance of the qualification as a prerequisite for the process validation is made clear in more recent guidelines on the subject (Appendix 15 to EU GMP Guideline entitled "Qualification and validation", EMEA "Note for Guidance on Process Validation" and ZLG Aide Mйmoire entitled "Inspection of qualification and validation in pharmaceutical manufacturing and quality control").
A minimum requirement before the process validation can start is successful completion of the OQ. The PQ data (long-term system-related performance test) are frequently obtained in tandem with the process validation during the first few months of routine production.
As nowadays all relevant process data is processed by computer, it is essential to demonstrate that these files can also be reliably "retrieved", i.e. in a form that can be read by human beings (see chapter 15 Documentation). Where no parallel recordings (paper printouts) of data acquired, transmitted and stored electronically have been made, the accuracy and integrity of the data cannot be subsequently checked. It is therefore necessary to perform computer validation for all manufacturing equipment using measuring and control technology or electronic data processing. Special aspects to consider are in this context access rights, software updates, incorrect input, influence of physical stresses (vibrations, heat, solvent vapours, dust, magnetic fields, etc.), archiving, speed of access to data etc. (See chapter 9 Computer Validation.) - Approved test procedure and validated analytical methods:
In order to assess correctly the test samples collected during the validation runs, the analytical methods used must be validated. If not, there is a risk, that a variability possibly observed during process validation runs cannot be traced back unambiguously - it might be caused by either the process or by variations in the analytics.
The test procedure must be approved by the designated responsible person if the validation batches are used as clinical trial samples, are stored for long-term stability studies, or - as is the case with concurrent validations - if they are to be used as commercial products. - Validated cleaning procedures:
If the validation batches are to be used as clinical trial samples or commercial products (concurrent validation), it must be ensured that patients are not endangered by contamination, i.e. from residual semi-finished products or cleaning agents or decomposition products. In this case, a validated cleaning procedure must exist at the time the process validation is carried out. If the validation batches are to be stored for long-term stability studies, a suitable cleaning process must exist as a minimum requirement to prevent the results being falsified due to contamination and subsequently being called into question. - GMP-compliant general conditions:
To carry out a process validation as an isolated measure would be pointless without qualified rooms, environmental monitoring, access control, sanitising, maintenance schedules or a functional documentation system. The training of all staff - not just the staff that carry out the validation - as well as functioning change control, documentation, calibration and maintenance systems are important prerequisites for the process validation. - Information flow:
Good communication between the departments involved, and also (if relevant) between contract giver and contract acceptor, is indispensable for the smooth running of validation projects. Generally, the validation experiences of the participants are very different - this applies particularly for (new) contractual partners. It is therefore necessary to agree on the terminology at an early stage and explain the basic procedure to all participants using the validation master plan. When carrying out the individual validation tasks, a good flow of information between the various interfaces will assist the timely achievement of objectives.
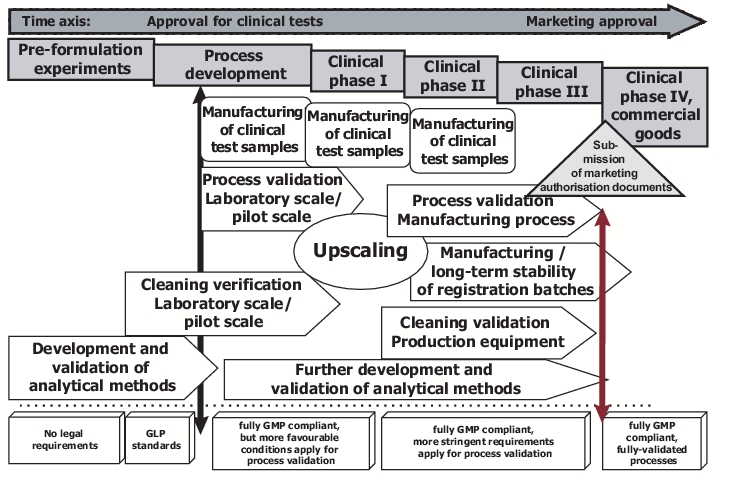
It will obviously not be possible to lay the foundations listed above shortly before the process validation is due to begin. Instead the various prerequisites for the validation must be fulfilled stepwise in tandem with the product development (see chapter 16 Research and Development). Figure 6 illustrates schematically the validation tasks that can be carried out at the individual development stages.
 |
4.1 What action should be taken if not all prerequisites have
yet been fulfilled?
For complex validation projects where (for example) a large number of tried-and-tested but very different preparations are manufactured using different facilities, bottlenecks will be inevitable as not everything can be qualified and validated simultaneously. To tell the truth, tasks such as qualification or cleaning validation, have not been finalised in many companies.
In these cases, the validation master plan has an important role to play: The overall validation requirements are listed in a "validation matrix" (see chapter 7.F.1 Validation matrix) and priorities are assigned to the individual sub-projects (bracketing may also be justified). In this case, an underlying rationale with respect to the assignment of priorities - such as product or process risk, production frequency, capacity of machines, experience with machines/facilities or error frequency (for example) - must be evident. This list of priorities must then be linked to a realistic schedule.
For the validation of a specific drug product, the status of the qualifications and cleaning validations must then be assessed and documented in the validation protocol as part of the risk analysis.
4.2 Manufacture of a development or pilot batch in the run-up
to a validation
If no data on process parameters acquired during development is available in case the process was transferred to a contract manufacturer, for example - one or several development batches (batch size approx. 3-10 kg, depending on dosage form and equipment) should be manufactured to obtain the missing data. In order to be able to estimate the transferability of the setting parameters to production equipment and future batch sizes, at least one pilot batch must then be manufactured (at least 1/10 of production batch size; pilot batches for veterinary drug products may be smaller if justifiable in accordance with EMEA "Note for Guidance on Process Validation"). If the process has not yet been optimised, one pilot batch is insufficient as the risks associated with the intended validation of a non-optimised process are extremely high. Ultimately, the following applies:
All processing steps must be controlled and critical processing steps must be validated.
|
Summary In order to be able to satisfactorily master a validation project in a timely and professional manner, sufficient resources should be made available. The management is fully responsible for ensuring that the validation is carried out properly - irrespective of whether this is delegated within a company or outsourced. The distribution of tasks and the responsibilities must be laid down in writing. The planning and practical implementation of validation projects is usually carried out by a team led by a validation manager. Care should be taken when assembling the validation team to ensure that all departments involved are represented. A process validation can only be successfully carried out if the necessary general framework is in place - for example: qualification, calibration and maintenance of equipment and facilities, cleaning validation, environmental monitoring, change control and training of staff. |
