|
Here you will find answers to the following questions:
|
1 Commissioning
|
Definition of commissioning: Commissioning describes the phase of a project during which equipment is procured. Once the detailed planning is complete, the procurement (or commissioning) phase follows during which the job is put out to tender, quotations are compared, decisions are made (comparability of quotations), the order is placed and final acceptance is carried out. |
The classic qualification activities take place in advance of the procurement process for the equipment. Responsibility for this logistical stage in the life cycle of the equipment is carried mainly by the technical and purchasing departments. However, a large number of interconnections exist between the areas of Good Engineering Practice (GEP) and applicable GMP requirements. This correlation is reflected by the contents of the DQ at the latest, and by evidence that all user requirements have also been translated into technical requirements from which the specifications have been derived. This may be clearly represented by using an implementation matrix (trace matrix).
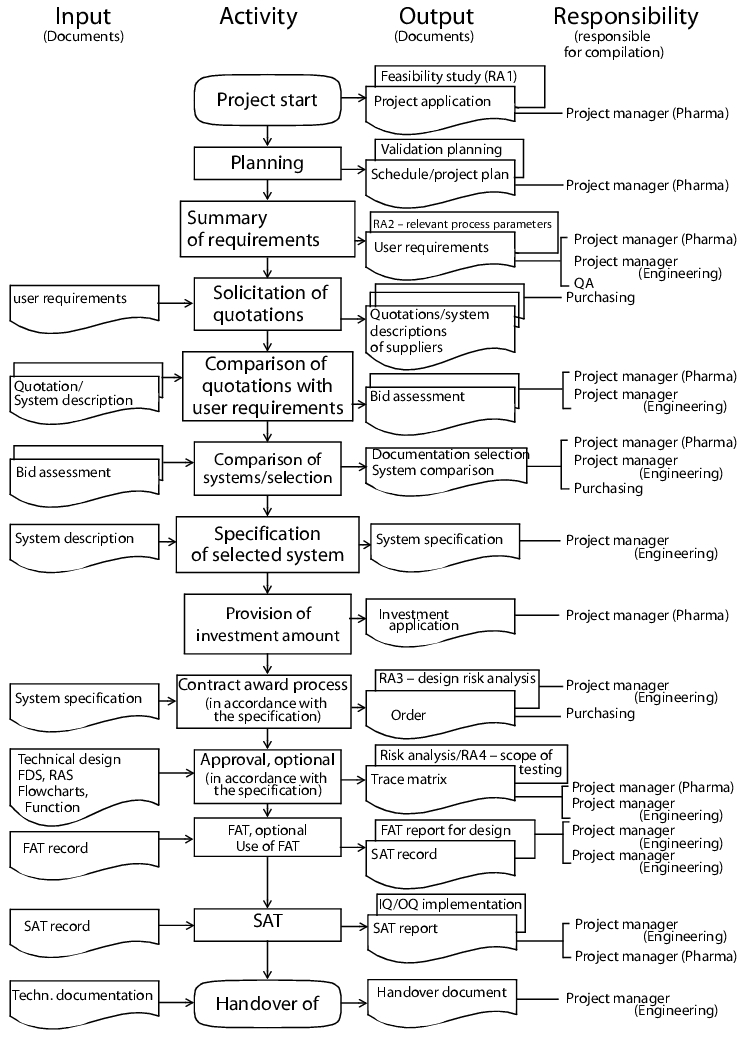
All project phases are shown in figure 1 - with particular emphasis on the procurement of accessories
 |
.
Project start (optional): At this point the technical and pharmaceutical project managers or persons responsible in the specialist department for the equipment to be procured are named.
Planning (optional): The activities plan (consisting of a schedule and project plan) is compiled in this phase. The implementation of preliminary trials and procurement of quotations with guide prices are also part of the planning. Market research and feasibility studies may also be included in the planning phase.
User requirements, user specifications: The operator defines the requirements at an early stage in relation to quality, quantity, demand and availability of the equipment. The technical department assists with the compilation of the user requirements, performs feasibility tests and amends the technical requirements. Following approval of the user requirements, the technical project manager coordinates all activities leading up to final acceptance. The user requirements are listed in the user specifications that subsequently play an important part in the qualification (also see chapter 6.D.1 User requirements (user specifications)).
Selection: The tender and processing of quotations, as well as negotiations in relation to the placing of orders, are carried out by the purchasing department based on the user requirements and market situation. In relation to all technical issues the technical project manager is the contact person for the equipment supplier. This person is responsible for compiling the documentation selection and comparing the user requirements with the system descriptions of the supplier (quotations). The documentation selection includes, as examples, the quotation evaluation (comparison of user requirements with quotation), the system comparison (comparison of individual quotation evaluations) and records of the contract award negotiations.
Detailed technical draft/system specification: The technical and pharmaceutical project managers compile the system specification (user specifications) for the pharmaceutical equipment based on the system description and quotation evaluation of the selected supplier. The contract is then awarded in accordance with the specification. The prerequisite for this is the approval and provision of the amount to be invested via the investment application. An important document for the subsequent qualification - the technical specification (see chapter 6.D.2 Technical specification) - is also compiled in this phase.
Trace matrix: Traceability is essentially characterised by a consistent referencing between user requirements, specifications and test cases, thus making it possible to follow cross references between the above elements (® traceability). The project manager is obliged to keep evidence of full implementation of the user requirements in technical specifications.
Technical design (optional): The technical design covers technical aspects of the equipment at such a level of detail that it can be used as the basis for development, manufacturing and installation. Examples of relevant documents are detailed drawings, bills of materials, assembly plans, P & I (piping and installation) diagrams, lists of components, functional descriptions and flow charts. Once these documents have been checked by specialists, manufacturing approval is granted.
Technical final acceptance tests: The FAT (Factory Acceptance Test) is the technical final acceptance test at the supplier's manufacturing premises. During this test, the completeness of components as well as materials, workmanship and quality are checked against the system specification and also, if required, the approved technical design. If significant defects are identified, the FAT is repeated at a later date. The equipment is not delivered in such an instance.
The SAT (Site Acceptance Test) is the technical final acceptance test that is carried out following delivery and installation. During this test, the correct installation of the equipment as well as all performance aspects, characteristics and functions are checked against the system specification and the approved technical design. If significant defects are identified, the SAT is repeated at a later date. The equipment is only brought into operation once the SAT has been successfully completed.
Safety tests that are mandatory - for pressurised containers and facilities in explosion-risk areas, for example - may also be carried out in addition to the SAT. The equipment is only brought into operation once the safety test has been completed successfully and it is subject to repeat tests throughout its entire life cycle.
FAT and SAT are not mandatory qualification documents, but they may be used if required, to support the qualification tasks by preventing the duplication of tests. The decision as to whether these final acceptances are carried out is made by the project managers. If FAT or SAT is omitted for standard equipment, the function test may alternatively be carried out as part of the qualification.
Handover to the operator: Once all final acceptances have been carried out, the equipment is handed over to the operator together with the manufacturer's technical documentation. All project documents that have been created or compiled by the technical project manager up to this point are also handed over to the operator.
The handover of these documents to the operator marks the end of the procurement phase and the start of the qualification phase for an item of equipment. It is of course possible for the qualification activities to be started beforehand, e.g. as early as the Factory Acceptance Tests. In particular, qualification activities that are outsourced to suppliers are not put on hold until the SAT has been completed. In such instances how the changes made to the facility are to be dealt with once the tests are underway must be defined.
2 Sequence
GMP or quality-relevant rooms, process facilities, machines, equipment and supply systems must be qualified. The term "equipment" is used in the following as a collective term. The qualification is carried out according to the steps shown in figure 2. The risk analyses are an integral part of the commissioning phase (cf. figure 1).
 |
Other formal assignment criteria are also possible. These must be precisely defined and any decisions for or against a sequence (e.g. in the qualification strategy) must be described. The design qualification may therefore be conducted as part of the installation qualification. The performance qualification may be assigned both to the qualification phase and the validation phase. The aforementioned basic procedure applies to the qualification of new facilities (prospective qualification). This procedure may need to be modified for the qualification of existing equipment ("retrospective" qualification) (see chapter 6.H.1 Retrospective qualification). When qualifying analytical equipment (e.g. HPLC facilities), the documented review of regular maintenance, calibration and system suitability tests may provide sufficient evidence of the qualified status of the equipment (see chapter 14.D Qualifying laboratory instruments).
An important component of all qualification activities is the risk analysis (see chapter 6 Risk analysis). During the risk analysis, potential risks are identified, their possible causes are investigated, and suitable measures for checking or minimising them are determined. The purpose of the risk analysis therefore is to determine the scope of the qualification and identify how resources should be used.
3 Qualification team
Qualification work is generally shared by a team. It is recommended, particularly when qualifying larger more complex equipment, that a qualification team be appointed using participants from the individual specialist areas of the project: e.g. production, development, technology, quality control, quality assurance, supplier, etc. The team formation may change during the various qualification phases.
The qualification team is led by a project manager (e.g. qualification representative) who should have sufficient competence in relation to the qualification project (see chapter 19.A Project management). In general, this role is fulfilled by specially-trained personnel (qualification engineers).
The qualification team carries out the following tasks:
- Compilation of the risk analysis
- Compilation of the qualification protocol
- Compilation of test plans, test packages and test cases
- Implementation and documentation of the qualification work
- Compilation of the qualification report
- Ensuring that other systems are implemented such as preventative maintenance, recalibration programme and standard operating procedures
4 Responsibilities
According to the EU GMP Guideline, the head of production and the head of quality control are responsible for the qualification of the equipment used in the relevant area in accordance with the latest scientific and technological developments (overall responsibility).
The qualification work may be delegated to competent staff - e.g. a qualification representative who in turn appoints a qualification team with the aforementioned responsibilities. In order to successfully carry out the qualification, it is important that this task is assigned the necessary importance at the company. Sponsorship arrangements, which are a feature of good project management, should be made in order to ensure that the necessary resources are made available.
5 Qualification by external service providers
In principle, a qualification package provided by the supplier or another external service provider may be used for the qualification. However, the specifications of the company's qualification policy in relation to content must be complied with. The qualifying company must specify what needs to be qualified and how this should be done even if it does not compile the qualifications itself. Responsibility for the qualification always remains with the manufacturer of the product and cannot be transferred to the supplier or service provider.
A service provider or supplier cannot approve the following documents: user requirements, user specifications, technical specifications, risk analyses, evidence of implementation (user requirements/user specifications/technical specification/test plans (trace matrices)), qualification protocols (DQ, IQ, OQ, PQ), test plans (IQ, OQ, PQ) and reports (IQ, OQ, PQ). Although a service provider or supplier may appear as the compiler on the cover of the document, the person who uses the equipment in his/her processes is ultimately responsible for ensuring that the contents of the document are correct and complete. On the cover of the document, a description should be included of the function of the individual signatories and the contents to which their signatures refer in order to avoid giving the impression that the qualification was determined by the service provider.
The following routes may be taken when outsourcing qualification activities. On the whole, in-house resources are not sufficient for the qualification of new facilities which means that nowadays, various combinations of the options listed are also used.
5.1 Integration of external capacities ("consultants")
into the qualification process
Although having a competent consultant on hand increases the capacity and know-how within a project, it must also be considered in such cases that this know-how will at some point no longer be available - i.e. when the qualification is concluded and the consultant's services are withdrawn.
It is important in such instances for the qualification team to rigorously engage the qualification strategy, the decisions made during the qualification and the contents in order to ensure an intensive transfer of know-how. This is the only way to ensure that the knowledge acquired during the qualification and its benefits can be used in the long-term and be available for inspections or requalifications. Intensive efforts must be made to ensure that the qualification team identifies with the qualification.
5.2 Transfer of parts of qualification activities to
consulting engineers
This approach frees up resources entirely as normally intensive training in the principles of qualification would need to be carried out at the authorised company's premises. When this arrangement is used, as well as the one that follows, the contract giver must have a competent in-house qualification consultant who manages these resources and deploys them correctly (qualification engineer). Here too, a subsequent loss of expertise must be considered in relation to this gain in resources.
5.3 Transfer of qualification activities to suppliers, acquisition
of qualification packages
The acquisition of qualification packages also increases the availability of internal resources. The supplier takes over components of the qualification, e.g. installation tests leading to as-built documentation (normally Good Engineering Practice), the implementation of specific function tests and accompanying documentation, initial calibration, etc. These kinds of services which include the risk analysis, evidence of implementation (trace matrix), design qualification, installation qualification, operational qualification and even the performance qualification and similar services, are provided as standard by suppliers for the pharmaceutical industry.
The following points must be observed when all-inclusive qualification packages are awarded with the order (see figure 3).
|
Contract giver/contract acceptor agreements |
|---|
|
Before qualification packages are ordered, an intensive analysis of the supplier's existing qualification strategy is required. A reconciliation of definitions and contents must be made to avoid the unnecessary duplication of work. Example: during risk analyses, checks must be carried out to determine whether it is a failure mode and effect analysis (FMEA) with Risk Priority Numbers (RPN) or a Hazard Analysis of Critical Control Points (HACCP). The type and scope must be precisely defined and laid down.
In order to determine the costs and ensure planning reliability, the qualification scope must be defined at an early stage together with the order. This requires a certain amount of experience as no functional descriptions/flow charts (also known as Functional Design Specification - FDS), valve control plans, programme structures, or similar are available at this point. A list of qualification requirements that describe around 95% of the qualification activities should be developed for regular orders. Therefore, analyses that are intended to verify the completeness of qualification packages are frequently difficult to perform.
The qualification packages and accompanying formats are standardised by suppliers and adaptations to individual qualification strategies must be negotiated. Today, the scope and depth of the qualification is generally determined by a risk analysis, trace matrix, or similar. This basis may only be developed once the order has been placed: depending on the circumstances this may lead to other expenditure and additional costs.
The maximum processing times for the approval of documents must be specified in the contract. The times allocated for the checking of test plans or similar are often too short. The checking of test protocols often requires several iterative steps that were not envisaged in the planning. As the first step, checks are carried out to determine whether the harmonised formats and specifications were complied with. Parallel checks are carried out to verify whether all contents of the user requirements and the risk analysis have been implemented in test protocols/test cases. To gain a clearer overview, these are carried out using implementation matrices (trace matrices). The initial content check is then carried out by the qualification team. Afterwards, it is often necessary to review the test plans for a first time. The reviewed plans are then checked in a second step (initial check to verify adoption of submitted corrections) and corrections are made to the semantics and elaboration of the test cases.
A check of the supplier's qualification resources should be carried out as project delays may lead to bottlenecks in resources. Suppliers also sometimes use external service providers resulting in additional interfaces in the coordination process. Generally in these cases the communication channels must be checked to ensure, for example, that agreements with qualification service providers are binding (additional costs and work).
It must be determined whether the supplier will only be compiling the test plans or whether the supplier will also be involved in the tests. Both options have benefits. The tests should be carried out by in-house personnel in order to achieve a maximum transfer of expertise between the supplier and the orderer. This could be linked with a training course for the operating and maintenance personnel. If resources are scarce and schedules are tight, it may be advisable to include the test implementation in the order, which means that only the test plans will need to be reviewed in-house. At this point, the allocation of roles for the tests should be agreed, e.g. how is a mutual check principle (four-eye principle) understood. However, the basic principle always applies that test plans may only be implemented once they have been approved.
The temporal and spatial sequence of the IQ/OQ tests must be arranged. This includes the agreement as to whether tests may be carried out directly at the supplier's premises (Factory Acceptance Test, FAT) and whether these may be assessed as IQ or part-OQ. In principle, these kinds of strategies must be described in the qualification protocol and possible risks assessed. One advantage of carrying out the IQ at the supplier's premises is, for example, that shortfalls in the documentation are easier to address and resolve. During the FAT, it is also feasible to test IQ/OQ test plans that have not yet been finally approved to guarantee a complete test run and check the test design for the last time.
The certificates required (filter, oils, material, seals, etc.) must be stated in the user requirements with the order as otherwise this may lead to delays during subsequent processing, and possibly culminate in disassembly of the materials used (seals). It is also sometimes necessary to define the contents of the certificates in order to ensure that, for example, material certificates such as 2.1B or 2.2B are sufficient for the qualification.
A binding project plan must be agreed that provides a detailed description of which documents must be available for approval and review and when. It is self-evident that milestones in the qualification also need to be secured contractually.
For orders placed abroad, the language to be used in the test plans must be specified in writing. As the verification of the tests is carried out at premises of the orderer, who will generally also want to use the test plans for change controls or requalifications, the orderer's language must be used.
It is expedient to check the test plans of previously completed projects or standard packages by the supplier in order to discuss layout and content issues prior to ordering. As a rule, it is desirable to include references to test documents in test descriptions and then of course append these to the test cases as raw data or reference them clearly. It is important that raw data (flow diagrams, function flow charts) that is used to carry out tests also carries inspection marks. Furthermore, use of the term "complete" in test protocols must be scrutinised as it is difficult to provide evidence of "completion". It is more useful precisely to limit the term towhere, for example, evidence is required only for all product-contact areas, or the test is carried out using lists that have been agreed in advance.
The rules for handling changes and deviations must be established. For example, how should these be integrated in the documentation, and when must the customer be informed?
6 Risk analysis
During the risk analysis, the potential risks to the process/product due to equipment (machines, systems, etc.) must be systematically investigated. A number of different approaches to the risk analysis are described in chapter 19.B Risk analysis.
 |
The aim of the risk analysis is to carry out a systematic examination of the equipment, design and process. This leads to the:
- Verification of the design
- Determination and evaluation of quality-relevant measuring points (e. g. specification of recalibration programme, operating ranges)
- Determination of maintenance activities (maintenance schedules and contents)
- Evaluation of the functionality of the automation system
- Determination of test contents for the qualification plans and addressing of critical points in the operating SOPs.
6.1 Risk analysis during the life cycle of a facility
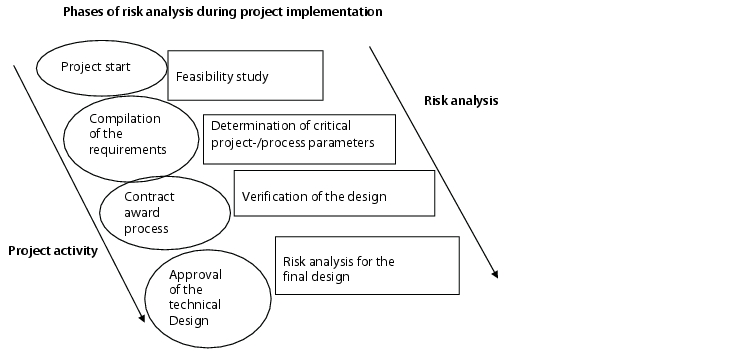
More than one risk analysis is carried out during the life cycle of the equipment (figure 4).
Ideally, the project should begin with a feasibility study that in a broader sense, also constitutes a risk analysis. Critical process parameters for the project/process should be determined as the project progresses, and particular attention should be paid to these during implementation, so leading to an optimum deployment of resources.
The next step should be carried out during the design phase in order to determine potential risks due to procedural changes, design changes, routing of pipes (arrangement of fittings, gradients, or similar) as well as the arrangement of the sensory equipment, and also have a timely effect on the design, the equipment, and the monitoring devices. This procedure is more difficult to achieve with equipment of prototype character, than with equipment that can be bought off the shelf. For prototypes, it is necessary for the project manager to involve the team in engineering questions and consultations to incorporate all aspects of the process in the design. Collaboration with the supplier can considerably increase the design quality of the equipment and the additional time required for this in the planning phase will be found worthwhile during the implementation phase.
If the final implementation planning for the equipment is available, the risk analysis to determine the scope of the qualification may be started. The evaluation of the equipment and specification of measures designed to minimise the determined risk is based on the drawings (P & I drawings, production drawings) and other specifications (function descriptions, hardware specifications). Possible measures in this case are:
- Checks during IQ, OQ and PQ tests
- Regular calibration
- Maintenance activities
- Addressing in standard operating procedures.
In order to trace that all measures have also been taken into consideration during the qualification, it is useful to produce evidence of completeness (trace matrix). To do this, references to the test protocol number, SOP numbers, maintenance schedules, or similar can be provided in the risk analysis.
6.2 Organisation of risk analysis
The risk analysis may be structured as follows (see figure 5).
The risk analysis is completed by adding a short description of the facility, a description of the procedure or reference to an SOP, a description of the implementation team, a tracing list for potential changes (problems detected or similar) and a final evaluation.
|
Example of the organisation of a risk analysis |
|---|
|
Cover with test, approval and change history
|
The risk analysis may be organised according to the following subject areas (figure 6).
|
Subject areas of the risk assessment |
|---|
|
The measures to be taken are specified in accordance with the determination of risk which may be achieved through the assignment of a risk priority number (RPN). The risk priority number (RPN) is a product of the following points:
- Probability of the occurrence of a risk (A),
- Significance of the risk/effect on the product (B),
- Detection of risk (E)
In processing terms, a higher priority number always takes precedence over a lower number. Each company must develop internal regulations in relation to the assignment of numbers and definitions for intended measures. These regulations might take the following form, for example. A risk is graded at least as average if it is affected by more than one factor - or the effect of the risk is only graded as high or low according to how the risk affects the process.
6.3 Implementation of the risk analysis
As risk analyses are very time consuming, good preparation and planning is recommended. To be able to answer all questions that arise sufficient available expertise is indispensable; it may be necessary to use the resources of the supplier. Following preparation, all components should have been entered in the template and a completeness check carried out. Intensive training of the team should be carried out beforehand to avoid discussions on procedural questions.
Intensive examination of the process and the equipment and therefore also the induction and integration of new team members is possible during the implementation of the risk analysis.
|
Risk analysis for equipment >XYZ< |
|||||
|---|---|---|---|---|---|
|
Equipment |
Function |
Failure |
Effect |
Evaluation of risk (ABE) |
Follow-up tracking of risk |
|
Specification of equipment components |
Specification of function |
Description of potential fault |
Description of effect of fault on product quality/safety at work |
Determination of the risk priority number |
Description of the measures for eliminating/minimising the risk |
|
Example |
|||||
|
Filter |
Particle reduction |
Filter |
Contamination |
1 x 4 x 5 = 20 |
Filter integrity test |
|
... |
|||||
Many companies offer "off the shelf" risk analyses which they have used to check the design of their equipment. In such instances it should be noted that, while the supplier almost certainly profits from the analysis, users may only share these benefits by checking the risk analysis against their own requirements and complete the process by orienting the analysis towards the specific use of their equipment. In order for the risk analysis to live up to expectations of an increase in expertise, it must be carried out separately by the user.
During a risk analysis, the potential risks are determined (in this case, for the equipment) - with regard to the effects on quality-relevant or safety-relevant functions, possible causes are investigated and suitable measures defined for checking or minimising these risks.
The purpose of the risk analysis is to carry out a systematic investigation of equipment including its hardware and software components and to derive measures that prevent or limit the identified risks. All potential risks with respect to the effects on product quality and safety, and possibly also operability, are determined and evaluated. The risk analysis is therefore an essential component of a qualification as it decides the content and scope of the qualification measures and is the basis used for the specification of quality-relevant measuring points and resulting calibration activities. In addition, the risk analysis may be used as the foundation for quality-relevant maintenance activities.
The risk analysis should be compiled at the earliest possible opportunity, ideally during the design qualification, in order to respond to potential risks through appropriate equipment design or specifications. This is more difficult to achieve with equipment of prototype character, than with equipment that can be bought off the shelf. In each case, an intensive examination of the topic of risk analysis is recommended at an early stage. Collaboration with the supplier can considerably increase the design quality of the equipment; the additional time required initially in the planning phase quickly pays off during the implementation phase.
The risk analysis is organised according to components, parts or functions of the equipment. Figure 7 shows the schematic layout of a risk analysis form.
Processing of the risks identified is carried out in accordance with the grading in terms of potential risk.
The basis for the implementation of a risk analysis might be the user manual and/or P & I diagrams as well as regulatory requirements, if required, as specifications for the functionality of the equipment under investigation.
As mentioned above, the scope of the qualification is derived from the risk analysis. This also means that a qualification does not always have to include the elements DQ - IQ - OQ - PQ and a reduced qualification scope can be quite justifiable in some cases. For example, a reduced qualification may consist of a risk analysis, supply scope analysis, calibration/maintenance and operating procedures (example: pH meter). The reasoning behind any reduction in the scope of the qualification must be provided via a risk analysis in every case.
Whatever method is applied, a comprehensive fault description may only be provided by the person who is familiar with the system (in this case, the equipment) and its individual processes/functions and who can develop a certain intuitive feel for typical scenarios.
|
Summary Qualification is necessary. Not only due to the regulatory requirements but also because any logical-thinking person will of course wish to ensure that equipment acquired for a considerable sum of money performs exactly as it should. Qualification requires teamwork. A working group is formed by representatives of various disciplines to carry out the qualification. Responsibility for the qualification remains with the head of manufacturing even if work is delegated to internal departments or external providers. Internal resources may be preserved by outsourcing qualification activities to consulting firms or acquiring qualification packages from suppliers. In so doing, the quality of the service as well as the subsequent loss of expertise must be considered. The risk analysis helps identify critical equipment parts and functions and therefore limits the qualification expenditure in all phases to quality-relevant aspects. Furthermore, the contents of operating procedures (SOP), maintenance activities and calibration programmes are defined by the risk analysis. |
