Qualification
Official requirements
|
Here you will find answers to the following questions:
|
1 Legal aspects of qualification
Qualification is an essential part of a pharmaceutical manufacturer's quality assurance system; it should demonstrate that facilities are suitable for their intended use and should also guarantee that the medicinal products are of an appropriate quality. Qualification is, thus, a basic factor for drug product safety.
Holders of manufacturing authorisations must have rooms and facilities at their disposal that are suitable for the production, analysis and storage of the relevant medicinal products. The suitability of facilities must be checked (qualification) where they are to be used for the production of medicinal products for which quality is decisive.
In the following, equipment and facilities are considered as a whole and all points raised apply to both, even if only "facilities" are referred to. An item of equipment is an object that is characterised by its internal technical processes. A facility is the sum of all equipment used for a common purpose.
The head of production is responsible for the necessary qualification measures within his scope of duties. Other company-internal departments (e. g. Engineering or Research and Development) or external service providers (e. g. consultants, contract giver for contract manufacturers) may be involved. The manufacturer (holder of the manufacturing authorisation) is responsible for ensuring that the necessary resources are available.
Operations and facilities are obliged to comply with the EU Good Manufacturing Practise guidelines for medicinal products (GMP Guideline). Regarding the "qualification of equipment", chapter 3.34 of the GMP Guideline states: "Manufacturing equipment should be designed, located and maintained to suit its intended purpose." Annex 15 to the EU GMP Guideline (see chapter 19 Tools for Quality Assurance) specifies how this requirement must be implemented.
While the EU GMP Guideline itself and its appendices are not legally binding, the Guideline does represent an interpretation of the basic principles laid down in the EU 2003/94 directive. According to Article 4, Paragraph 1 of this directive, the manufacturer must ensure the conformance of all manufacturing operations with good manufacturing practice. According to Article 8, Paragraph 3 of the directive, premises and equipment must be checked to make sure they are suitable for carrying out critical manufacturing operations where product quality is concerned. (Qualification).
Member states have implemented the directive as national law. The requirements of the EU GMP Guideline and its appendices reflect state-of-the-art technology. In particular, the suitability of rooms and facilities as well as a guarantee that state-of-the-art production and analysis will be carried out are important prerequisites for the issue of a manufacturing authorisation. If the manufacturer is not able to demonstrate the suitability of facilities and equipment, the manufacturing authorisation may be withheld or withdrawn.
As only the principles of qualification and validation are shown in Annex 15 to the EU GMP Guideline, PIC/S document PI 006 entitled Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation may be used as an interpretation and implementation aid (chapter F.1 Recommendations on Validation Master Plan Installation and Operational Qualification Non-Sterile Process Validation Cleaning Validation (PIC/S PI 006)).
This document applies primarily to investigators in the PIC/S member states for whom it is intended as instructions for preparing an inspection and as an advanced training aid for qualification/validation. As, from the PIC/S viewpoint, this reflects the latest scientific and technological developments, valuable information may be found here for the industry regarding the implementation of the specifications in Annex 15.
The objective of all qualification work is always to verify the suitability of the facility or equipment for its intended purpose within defined limits, otherwise known as acceptance criteria. It follows that acceptance criteria must be defined before the qualification is carried out as they are core elements of every qualification plan. Requirements arising from batch production records, registration documentation, standards or risk analyses (as examples), may be referred to when setting up acceptance criteria.
If implementation of the qualification is assigned to third parties, responsibilities must be clearly defined. A suitable contract has to be drawn up that sets out the tasks and responsibilities of each party in writing. The head of production must make sure that qualification work is carried out based on the latest scientific and technological developments. The responsibility for compliance with drug-related legislation is not transferred with tasks when they are assigned but remains with the head of production.
There are many reasons why companies often use external services when carrying out qualification. Frequently there is insufficient in-house knowledge and experience or not enough personnel to carry out a state-of-the-art qualification project. This is a typical field for consultants whose activities range from providing one-off consultations through to full implementation of the qualification and accompanying documentation.
While the buying in of external expertise is permissible, and is also useful in many cases, it is often wrongly assumed that responsibility for the work can also be transferred to third parties as well as the work itself. Chapter 2.5.11 of PIC/S document PI 006 therefore expressly states that the contract giver is ultimately responsible for proper implementation of the validation work: "In such cases, the responsibility lies with the contract giver to ensure that the required standards of the quality of the work which is carried out, for programme control and for documentation are met."
In this context, a regulation in the American legal system is also drawn to your attention. Paragraph 211.34 of the Code of Federal Regulations (CFR) states: "Consultants advising on the manufacture, processing, packing, or holding of drug products shall have sufficient education, training and experience or any combination thereof, to advise on the subject for which they are retained. Records shall be maintained stating the name, address and qualifications of any consultants and the type of service they provide" (chapter D.1 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceuticals).
Analogous to the regulations for the qualification of contract manufacturers and suppliers, pharmaceutical manufacturers should therefore also make sure at the outset that external consultants/service providers are capable of undertaking and documenting qualification work in accordance with the latest scientific and technological developments. The distribution of tasks and responsibilities for both parties should be described in a contract.
2 Documentation of the qualification
The GMP Guidelines for documentation apply in general for the layout and compilation of qualification documents (chapter 15.A Official requirements and chapter 15.B GMP-conforming documentation). The plans at all qualification stages must be authorised by the head of production and quality assurance.
The documentation should be retained for at least five years once the facility or equipment has been shut down. According to Annex 15, No. 2 of the EU GMP Guideline, a company's current qualification projects must be described in a validation master plan (chapter 7.F Validation master plan).
This is used by the manufacturer to determine the relevant basic principles and procedures for the qualification. It also defines the qualification deadlines and it may be used to estimate the necessary resources. Additionally, the validation master plan enables the GMP inspector to understand how the company approaches the qualification, and how the necessary activities are defined and organised.
Protocols must be used to define the detailed qualification regulations for each facility (see chapter 6.C Qualification documentation, chapter 6.E.1 Examples of IQ plans and chapter 6.F.1 Examples of OQ plans) and these must be authorised by the persons responsible.
The implementation of the individual qualification projects should be documented in corresponding reports according to the specifications of the qualification plan used as the basis. Subsequent deviations from the qualification plan, and particularly changes to the acceptance criteria or the test procedure, must be well founded and formally authorised. A qualification report always ends with a conclusion regarding the suitability of the facility concerned.
Once the qualification has been successfully completed, approval must be given in writing in order for the facility to be used in routine production. This approval may be granted by including the signature of the head of production in the qualification documentation.
3 Design Qualification (DQ)
The qualification of facilities and equipment is part of their life cycle (figure 1).
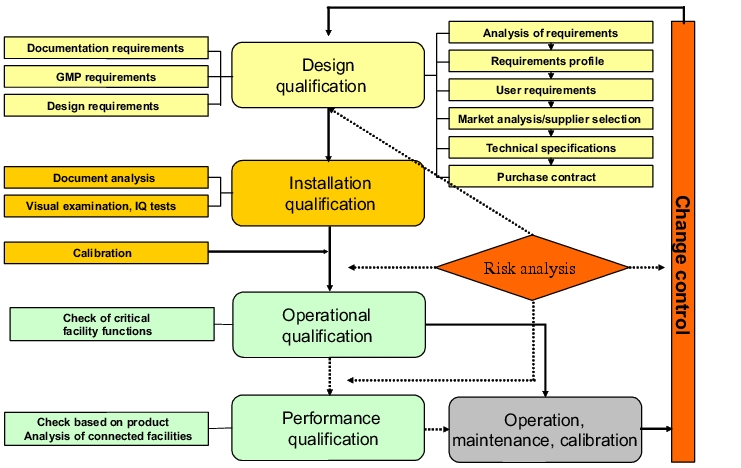 |
The first stage of a qualification should be the design qualification (DQ). According to Annex 15, conformance of the design with the GMP requirements should be demonstrated and documented.
The design qualification encompasses the documentation of the planning phase including the decision as to which facility will be used. The DQ should define the requirements for the intended facility.
The requirements of the contract giver for the scope of supply and services (cf. figure 2) (user requirements) or the agreement with the contract acceptor regarding implementation and handling of the project (technical specification) are described in the design qualification documents.
|
Contents of the design qualification (DQ) |
|---|
|
1. Purpose of the equipment/facility 2. Legal requirements (laws, regulations, standards) 3. Design requirements
4. Requirements for the installation
5. Performance data
6. Facilities for control, regulation and measurement 7. Requirements for maintenance, cleaning and care 8. Evidence of the functionality of the equipment/facility, e. g. records of trial runs at the premises of the facility manufacturer (final acceptance documents) 9. Requirements for the accompanying documentation
10. Customer service 11. Necessary supplementations and modifications following award of contract 12. Installation schedule at premises of contract giver |
Before acquiring a new facility, the manufacturer must first determine the requirements (processes, products, capacity) that must be catered for. A number of different areas at the company are usually involved in the assessment of requirements that may be used to derive an informal requirements profile for the new facility. This requirement profile is transferred to the user requirements. The requirements of users including all limit conditions must be described in the user requirements. It should be possible to qualify and test these requirements. The user specifications describe the economic, technical and organisational expectations of the contract giver in relation to a facility and they also define the objectives and purpose of the facility.
When compiling the user requirements, a differentiation should be made between essential (compulsory) requirements and desirable (optional) requirements. In practice, it will not be possible to implement all the ideas of the future operator. Whether a facility manufacturer for a specific facility can be found and whether he is capable of delivering a suitable quality always depends on the market situation. A thorough market analysis will therefore determine what offers are available. It is quite possible that changes to the user requirements may be required as a result of such an analysis as new aspects in relation to the design of the facility may come to light that could not be taken into account when the user requirements were compiled.
Not all suppliers in the market will be in a position to guarantee that the facility will be designed and produced according to the principles of Good Engineering Practice (GEP) as defined in the standards and guidelines (baselines) of the ISPE (International Society for Pharmaceutical Engineering): "Established engineering methods and standards that are applied throughout a project's 'life cycle' to deliver appropriate, cost-effective solutions". When selecting a suitable supplier, checks should not only be made to establish whether the manufacturer actually supplies the required facility, but also to determine whether the manufacturer can provide evidence that he is able to comply with the aforementioned principles of GEP. This check must be carried out as part of a supplier qualification and should also consider aspects of service such as support during qualification, instruction of operators and maintenance.
The user requirements are dispatched to suitable suppliers following the supplier qualification (chapter 18.G Inspection of suppliers). The suppliers compile a technical specification on the basis of the user requirements and forward this to the prospective customer as part of a quotation for the conclusion of a purchase contract.
The user requirements are detailed in the technical specification and the implementation requirements described in an extension with reference to specific approaches. A definition is given in the technical specification as to how the requirements are to be implemented and what is to be used to achieve this.
Basically, the design is determined to be qualified if the technical specification corresponds with the user requirements. A documented comparison of the technical specification by the manufacturer/supplier of the facility with the user requirements of the pharmaceutical manufacturer should demonstrate that at least the compulsory requirements have been satisfied.
Before the purchase contract is concluded, or before the facility is delivered, it may be necessary to make sure that the user requirements are complied with at the manufacturer's premises (Factory Acceptance Test, FAT - "The partial commissioning and qualification of equipment and/or systems prior to their shipment from the fabricator's site", ISPE). To do this, a visual inspection of the facility is carried out at the manufacturing site. If required, and if circumstances permit, suitable test runs are carried out.
4 Installation Qualification (IQ)
The correct implementation of the aforementioned requirements when assembling/setting up the facility is documented in the installation qualification (IQ) (cf. figure 3).
The check of the installation is generally carried out on the basis of the requirements previously established during the design qualification.
The installation qualification should be carried out on new or modified buildings, systems and equipment as defined in the glossary of Annex 15: "The documented verification that the facilities, systems and equipment, as installed or modified, comply with the approved design and the manufacturerґs recommendations".
The contents of figure 3 constitute the minimum requirement for the scope of the installation qualification.
|
Installation qualification according to Annex 15 |
|---|
|
The installation qualification serves as a check of the documents that were required for the design qualification. These include the drawing of the facility, lists of settings, lists of components, instruction manuals, operating instructions, circuit diagrams, spare parts lists/wear parts list, maintenance and cleaning procedures, certificates (material, CE etc.), calibration documents, software and hardware documents and a list of product-contact parts (surfaces and materials). The documents must be correct, complete and up to date.
A visual examination is also carried out of the components delivered, to ensure fault-free workmanship, correct assembly and set-up, correct implementation of all utility connections, and connections with upstream and downstream machines.
The individual working steps should be carried out using inspection or work sheets (with a uniform layout) and the acceptance criteria established at the design qualification stage as a foundation.
All deviations or changes identified during the installation qualification must be documented to be used as the basis for assessing the need for the compilation of a defects list (including responsibilities and deadlines). The results are then listed in an appropriate form in the final report and comprehensively checked once again.
If it can be demonstrated that the installation of the facility corresponds with the relevant specifications, the installation qualification may be finalised with the signatures of the responsible persons.
5 Operational Qualification (OQ)
The Operational Qualification (OQ) provides evidence that the facility is functioning on the basis of established parameters and within defined limits. The OQ is a testing process and therefore the test methods to be applied, as well as the acceptance criteria, must be defined and laid down in advance.
Annex 15 states that the installation qualification should be followed by the operational qualification and that the minimum scope of the operational qualification is based on knowledge of the process, systems, or the facility, and also tests that facilitate the definition of an upper and lower operating range. These tests should be carried out under worst-case conditions.
As no release is required for the installation qualification, this could be approved together with the operational qualification. For logical reasons, the operational qualification should only be started if no significant (quality-relevant) defects were found during the installation qualification.
The basis of the operational qualification is a risk analysis during which the critical parameters of the facility and the environmental conditions are assessed. All relevant measuring equipment must be properly calibrated prior to the operational qualification. The appropriate final acceptances for safety-relevant components/facilities should have been carried out. A preliminary draft of all instructions necessary for the operation of the facility should exist as a minimum requirement.
A practical test phase then follows during which the various functions are checked in detail. Depending on the facility, the environmental conditions may need to be checked. The implementation of the cleaning/disinfection and maintenance work should also be checked at this stage.
To check the functionality, a sufficient number of suitable test runs must be carried out based on predefined qualification protocols for the operational qualification. The implementation of these tests must be recorded in qualification reports (cf. figure 4).
|
Qualification protocol (OQ) |
Qualification report (OQ) |
|---|---|
|
|
Efforts should be made to ensure that the conditions under which this test phase is carried out are as close to reality as possible, i. e. routine manufacturing conditions. The same personnel that will subsequently be responsible for operating the facility should be present at this trial run.
There are fundamental problems associated with the adoption of FAT test results for the OQ phase. If functional tests have already been carried out at the premises of the facility manufacturer as part of the FAT test (cf. chapter 3 Design Qualification (DQ)), these test results may only be adopted for the OQ phase if it can be proved that the test conditions at the facility manufacturer's premises are transferable to the operations of the pharmaceutical manufacturer (e. g. identical installation, test under realistic conditions) and that the test was based on an approved qualification protocol as shown in figure 4.
The operational qualification ends with a formal release which should also conclude the calibration and training as well as the final versions of the operating, cleaning and preventative maintenance instructions.
6 Performance Qualification (PQ)
Once the operational qualification has been carried out, the check of the facility's suitability is generally complete; though it may still be necessary to carry out a performance qualification (PQ) for specific products or for facilities that are linked to systems. Annex 15 defines performance qualification as, "The documented verification that the facilities, systems and equipment, as connected together, can perform effectively and reproducibly, based on the approved process method and product specification." Therefore, the performance qualification, although more facility oriented, is a test procedure that from the beginning is concerned with individual processes. If systems are created through the assembly of individual equipment that is already qualified, the functionality of the overall system (as connected together) must also be checked. Generally, the performance qualification should only start once the installation qualification and operational qualification have been successfully concluded. In individual cases it may be logical to combine elements of the operational qualification with the performance qualification. According to Annex 15, the tests carried out during the performance qualification must use production material, or suitable alternatives; also, tests that incorporate the upper and lower operating ranges must be included.
A fundamental part of the PQ is performance testing of the facility with all production materials subsequently processed during routine operation. During this testing, it should be observed in particular whether specific materials have an effect on the performance criteria of the facility due to their individual properties. Where the bracketing principle (chapter 7.F Validation master plan) is applied during the risk analysis, all other materials used must be taken into account in addition to starting materials and packaging materials.
If the product properties in question are simulated when the performance qualification is carried out ("simulated product"), all possible product categories must be taken into account.
Upper and lower limits for production on the facility should ultimately be defined with reference to the respective criteria to be observed (moulding pressure, temperature, etc.) for all materials processed.
As is the case with all other qualification phases, the PQ implementation must also be documented in a report. This report may be part of the documentation for the operational qualification, or the process validation, or it may be handled separately within the documentation - there are no specific requirements. The PQ should, however, be recognisable as such in the documentation.
It must be possible on the basis of the report to clearly identify which materials may be processed on the facility and under what conditions.
As with the operational qualification, deviations from the anticipated results and their consequences (e. g. repeat control testing of the type and scope of supply connections and waste outputs) must be precisely noted during the performance qualification.
The transition to the process validation may only be made once the questions and problems arising in relation to the PQ have been clarified satisfactorily in the estimation of the persons responsible.
Tests that are carried out during the PQ may also be used for process validation provided that they offer an insight into the safety/suitability of the process. However, it is generally not possible completely to dispense with a process validation on the basis of the performance qualification results.
7 Qualification of established facilities
For the qualification of established (in-use) facilities, systems and equipment (according to Annex 15), sufficient data must be available in order to carry out a subsequent assessment and review of the various critical operational parameters and associated limits. The directions and records for calibration, cleaning, preventive maintenance, operating instructions and personnel training should also be documented.
The qualification of established facilities should comply with the life cycle model shown in figure 5. All qualification phases must be implemented on the basis of qualification protocols that have been approved beforehand. The DQ phase starts with a design check of the facility in its current state as described in chapter 3 Design Qualification (DQ) because established facilities must also be checked to determine whether they still meet the applicable minimum requirements.
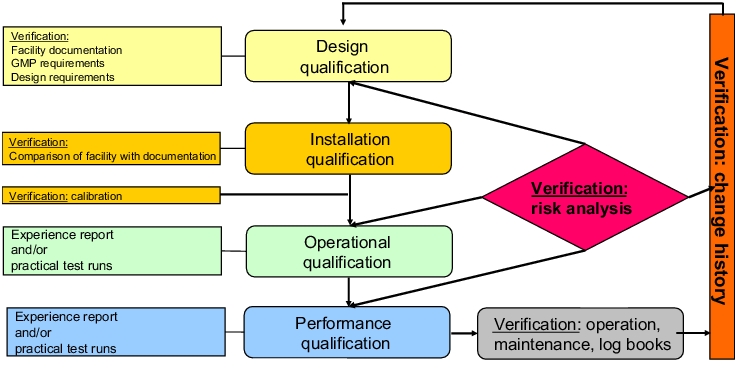 |
In the IQ phase for established facilities, an inventory is made of the installed components and existing documents (cf. figure 3). Without a detailed and up-to-date account of the current condition of the facility (as-built), a check regarding suitability and subsequent change control will not be possible.
The check carried out during the OQ phase that follows the IQ phase to verify that the facility is functioning properly may be based either partially or completely on existing data obtained from an experience report. In an experience report, reliable data is compiled and evaluated for a defined period with the objective of checking whether the facility in question is functioning properly using a qualification protocol as the basis. For data sources (cf. figure 6), only evidence that can be verified as accurate and that is based on facts obtained through observation, measurement, investigation or other ascertainment methods may be used (cf. DIN ISO 8402:1995).
|
Data sources for an experience report |
|---|
|
It must also be decided, on the basis of a risk analysis, which critical facility parameters must be covered by the experience report. For the compilation of suitable data for these parameters a production period must be selected during which, in general, no significant changes were made to the facility. The facility status during this period must also correspond with the current status of the facility to ensure transferability of the data evaluation to the actual status. The experience report should not be limited to a compilation of data, it should always be accompanied by a statistical evaluation (e. g. quality control chart analyses, machine capability studies).
When compiling the experience report if it is determined that the above requirements in relation to data sources cannot be met, or can only partially be achieved, additional practical tests for critical parameters may be carried out for which, due to the data situation, a retrospective evaluation is not permitted. A purely retrospective data evaluation is not permitted if the raw data that forms the basis of this evaluation is missing; if the data does not cover the current range of operational parameters; if major changes to the facility have been made; or if deviations or trends in the function of the facility have emerged during the period under observation and have not been clarified.
8 Requalification
The qualification does not consist of one-off activities such as the introduction of a new manufacturing facility. The initial implementation should always be pursued as a continuous programme (cf. PIC/S document PI 006, Chapter 2.5.12).
Qualified facilities are subject to a range of influences that may endanger the qualification status:
- Wear and tear and/or insufficient maintenance work
- Calibration intervals too great
- Misuse/incorrect use of facilities
- Accumulation of minor changes having a critical overall effect
- Process instructions not followed correctly
- Change of general conditions (laws, GMP requirements)
Facilities should therefore be evaluated at specific intervals to ensure that they remain in a qualified state.
The documentation requirements for requalification are the same as those for initial qualification and therefore similar documents may be used in many cases (cf. PIC/S document PI 006, Chapter 6.6.3).
"Where no significant changes have been made to the validated status, a review with evidence that facilities, systems, equipment and processes meet the prescribed requirements fulfils the need for revalidation." (cf. Annex 15, No. 45 to EU GMP Guideline)
There is no general rule stating how often (i. e. the frequency at which) facilities should be requalified. It is impossible to state categorically when and how qualifications should be carried out as the size and complexity of production processes and facilities vary considerably (cf. PIC/S document PI 006, chapter 2.5.5). The specification of a suitable requalification deadline therefore clearly depends on the risk evaluation by the pharmaceutical manufacturer. The extent to which conclusions may be made regarding the occurrence of defects, their frequency and probability of detection depends on the type and scope of records that provide an insight into the function of the facility. In this regard, all manufacturers are at an advantage who have already introduced continuous monitoring of all critical facility functions (as is standard practice in other safety-relevant areas of industry). This check may also be carried out using quality control cards and machine capability studies (to determine CMK value).
For change projects where only one specific aspect has been modified, it is not absolutely necessary to requalify a facility from scratch. However, it is important to carefully assess the type of change in order to identify possible consequences and to establish the precise scope of the requalification.
|
Summary Qualifications should document the suitability of facilities. The head of production is responsible for the qualification of manufacturing facilities. When transferring qualification work to third parties, the tasks and responsibilities must be defined in a contract. Qualifications must be documented in the form of qualification protocols and reports. The qualification must be implemented in stages: DQ, IQ, OQ and (if required) PQ. Precise instructions of the work involved at each stage must be given (qualification protocol) and the implementation of this work must be authorised. The qualification of old facilities is feasible provided a detailed experience report can be compiled. Facilities that have been successfully qualified must be released for routine production. Facilities should be evaluated at specific intervals to determine either that they are still in a qualified state or that requalification should be carried out. |
