|
A key
component to successful process validation of synthetic chemical
processes for the manufacture of Active Pharmaceutical Ingredients
(APIs) is developing a comprehensive validation program. This paper
describes an approach to establishing such a program starting with a
corporate policy through the various components of the validation
exercise. A lifecycle approach to the validation concept is
critical, and begins with the development program and does not end
until the product is retired. Circumstances that are unique to
synthetic chemical pathways are presented. The paper describes
specific details and examples for establishing validation master
plans, validation protocols, and writing the final validation
report. Additionally, other topics such as deviations and failures,
homogeneity, shipping, and process trending are
discussed.
The
paper describes activities that relate directly to manufacturing and
quality assurance components of such a program. However, the paper
does not attempt to deal with a corresponding program in analytical
methods since this is a subject of its own. Additionally, the paper
does not deal with any detailed aspects of equipment cleaning
validation for the same reason as described above for analytical
methods. Equipment cleaning methods should be developed in situ with
chemical process development, and cleaning validation should occur
either prior to or concurrent with process validation.
Definitions
Active
Pharmaceutical Ingredient (API): A substance intended for use as an
active ingredient in a finished dosage form of a drug. Such a
substance is intended to furnish pharmacological activity in either
the diagnosis, cure mitigation, treatment, or prevention of a
disease.1
Intermediate: A compound that is produced en route to
an API. The compound has all or at least a portion of its structure
that is incorporated into the structure of the final API.
Development Report: A report, or series of reports, that
describe in detail the development history of an API or
intermediate. The report should include a summary of all laboratory
experiments used to support the current process, and include a
comprehensive batch history profile with a summary of all process
changes.
Policy:
A written ideology or philosophy concerning a certain subject, and
the basis for which procedures are established.
Procedure: A written set of instructions on how to perform a
specific task, e.g., Standard Operating Procedures
(SOPs).
Quality
Assurance (QA): An independent unit that reviews documentation and
activities for compliance to Current Good Manufacturing Practice
(cGMP).
Quality
Control (QC): An independent unit that performs laboratory testing
on a compound using prescribed methods. Included in this definition
are groups that are responsible for developing and validating test
methods, performing routine release testing, and maintaining
stability programs.
Critical Parameter: A processing parameter that has a
critical effect on the downstream processing or quality profile of
the intended product. If the parameter is not tightly controlled,
the downstream process and/or quality profile of the product could
be negatively impacted.
Introduction
Process
validation is arguably the most important event that occurs during a
product’s process lifetime. Too many companies view process
validation as a tedious event that takes time, money, effort, and is
only needed to satisfy the FDA. Although born as a result of
regulatory considerations, process validation should be viewed as a
scientific event. That is, process validation is a demonstration and
verification of the science that went into developing a process, and
should be viewed as a significant accomplishment in the science and
art of process development.
What is
process development? It is the documented demonstration that all of
the effort that went into developing a process has led to a process
that will consistently produce a given product. This means that
process validation does not begin with the first batch to be
validated, or the protocol, or the master plan. Rather, it begins in
the laboratory at the earliest stages of process development, and is
a continuous event that follows a process throughout its lifetime.
When viewed in this fashion, and developed in a proactive,
comprehensive manner, one has established a program that supports
the quality and success of the product. This paper will describe an
approach on how to develop this type of program.
Does
this mean that, if performed correctly, such a program will
guarantee problem-free processing for the life of the product?
Certainly not. Humans may like to believe that they can master
nature, but nature shows us all too often that we are mistaken.
Simple or complex chemical processes all have the ability to go
their own path, albeit, many times with the helping hand of humans.
This paper will also describe approaches to dealing with problems
that may be encountered during process validation.
The
Validation Program
A
successful validation program is comprised of many components, many
of which need to be implemented well in advance of the actual
validation exercise itself. Using a sports analogy, the success of
the performance (i.e., validation execution) depends upon the effort
and efficiency that went into preparing for the event (i.e.,
preparation and training). Therefore, a successful validation
program begins at the very early stages of process development
itself, and does not culminate with the validation event, but rather
when the process eventually retires. It is a process lifetime
event.
A validation program is comprised of many components
including the written aspects (policies, procedures, protocols,
etc.), the personnel (departments, technical experts, consultants,
etc.), and activities (qualification, training, etc.). All of these
components should be described and coordinated into a functional,
comprehensive program.
Figure
1 depicts a typical hierarchical approach to such a program through
a program pyramid. The view can be expressed that the top levels of
the hierarchy govern the program, but the foundation to success is
in the science that is put into process development, equipment and
facility qualifications, analytical method development, etc. and
ultimately in personnel expertise and training.
Corporate Validation Policies
A comprehensive corporate validation program is assembled
beginning at the very top with a well-defined validation policy. The
policy should outline the organization’s general validation
expectations over the lifetime of the process. The policy should
define the scientific expectations and documentation that begins
from the first day of process development. Some examples of these
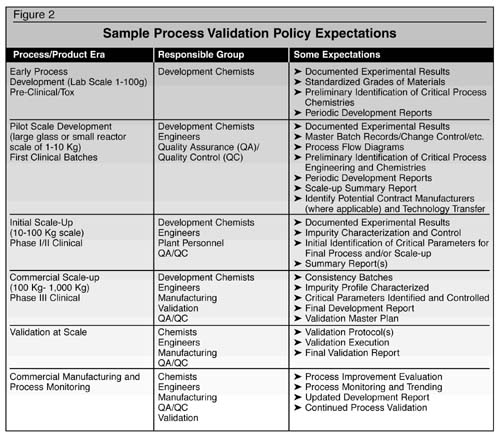
expectations are described in Figure 2.
An important aspect of
such a global policy is that it exposes employees, who may have very
special tasks, to the broader validation program. The employees then
become better trained and cognizant of their role in the validation
program. For example, chemists who may be normally focused on
process development will do much better in organizing their data,
writing reports, etc. if they understand what purpose their data and
conclusions will serve in the end when process validation protocols
are assembled and executed. General training in the concepts of
process validation should be given to all individuals who are part
of the development and manufacturing program.
Another
important feature of the policy should be the participation of
development personnel in the actual validation exercise. Development
personnel are generally the most knowledgeable concerning the
process, and can ensure that the technology has been appropriately
transferred into the commercial manufacturing environment. In
addition, the experience of participation is invaluable to the
chemist and will help them during the development phase.
Finally, the policy should describe the responsibility of
maintaining the product/process during the product lifetime
(lifecycle approach). Each organization may have a different
infrastructure that is used to accomplish this. The policy should
describe the responsibility for monitoring and trending, process
improvements, change control, etc.
Corporate Procedures Related to Process
Evaluation
In
today’s world, it can be said with a high degree of certainty that
most API processes are multi-step synthetic pathways that generally
may involve at least one complex chemical transformation. A
reasonably accepted validation axiom is that the final process step
that produces an API (and any purification thereof) needs to be
validated. However, a debate can be started as to when to perform
process validation when intermediates are involved. Therefore, an
approach to an evaluation procedure for intermediates will be
suggested.
There
is no simple answer to the question of when to apply process
validation for intermediate steps in a multi-step process. Each
process must be independently evaluated. However, this does not mean
that a coherent policy or procedure cannot be established that
describes how that evaluation process should take place. In fact,
putting such a procedure in place only benefits those that are
trying to conduct the evaluation. How should the question of when to
validate intermediate process steps be approached in a
procedure?
Even
though each process needs to be evaluated independently, there are
some common factors and criteria that can be applied when conducting
an evaluation. Multi-step synthetic processes can be classified at
the simplest level as either being linear or convergent, or a
combination of both approaches. In a linear synthesis, a starting
material is either built upon, re-arranged, or further elaborated
through chemical processes to eventually achieve the desired
molecular structure. A convergent synthesis, on the other hand,
builds pieces of the molecular structure independently, and then
assembles the pieces to achieve the desired molecular structure.
Many processes incorporate both elements.
The
first step of the evaluation is to determine what the starting
material is, what are isolated and non-isolated intermediates, what
is (or are) the ultimate intermediate (or final intermediate), and
what synthetic scheme will be required to produce a final API. In
fact, determining what constitutes a starting material is a
debatable issue.
This
author has generally taken the following approach in evaluating
whether a material is a starting material. First, the material
should be a readily available item that has a standard grade (or
grades) associated with it, and has been well characterized. It may
be produced under contract by a vendor, but in that case, it should
be produced by a known and established process, and the end product
should be, again, capable of achieving a standard grade. Second, the
vendor should be qualified (under a vendor qualification program) so
to ensure that a material meets consistent quality standards. A
rudimentary quality agreement should be established to outline
change notification and quality requirements.
If a
material is manufactured by a contract party and the process was
supplied by the innovator, the material is simply a third-party
manufactured intermediate. The contractor now becomes an extension
of the innovator, and the transferred process must be included in
the full validation evaluation.
Once a
starting material has been established, each intermediate needs to
be evaluated in two ways:
- How
it contributes to the final API, particularly in regard to the
impurity profile, and
- The
specific process for that intermediate. Although isolated
intermediates should be fully evaluated, non-isolated
intermediates should also be evaluated, since there may be
processes where stricter controls may be required.
For
example, an intermediate may not be isolated due to structural
instability when not in solution. In this case, concentration and/or
potency of the intermediate may be a critical attribute that needs
to be controlled.
During
development stages, an emerging impurity profile of the API should
emerge. This profile should be split into two separate profiles to
include the final purified API and the initial crude API. From the
crude API, an impurity fingerprint can be established concerning the
process. Questions can be asked such as what intermediate process
steps may contribute to the crude profile? What are the boundaries
for purification to the final API? What should be the desired
quality attribute of intermediate A in order to achieve the desired
end product? This is why it is important to characterize process
impurities as early in the development process as
possible.
From
this type of evaluation, each intermediate process step can be
evaluated as to its relative importance in the end product. If an
intermediate needs to be controlled since it could contribute to an
adverse quality profile of the API, process validation should be
applied.
Another
reason to employ process validation is if an intermediate step is
particularly difficult to control or is technically difficult.
Process validation in these cases would help ensure that the
manufacture of the intermediate is consistent and is performed with
an appropriate level of attention.
An
important factor in approaching any process validation evaluation is
to avoid considering it only a regulatory requirement to which only
a minimal effort should be applied to satisfy the regulatory
requirement. Rather, process validation should be viewed as a
scientific approach to ensure critical processes have the integrity
required to make a quality product. Additionally, process validation
makes good business sense in that well-developed and validated
processes ultimately save time and money and increase quality, which
is accomplished by reducing, although rarely eliminating, costly
batch failures, investigations, inappropriate or inefficient
processing, constant reprocessing, etc.
Finally, there has always been some discussion whether the
validation exercise should be designed so as to “stress” the
accepted parameters by operating at the maximum or minimum accepted
level, or to aim for the target of each parameter. Some companies
have attempted to matrix the parameter limits in an effort to test
the extreme limits of process parameters. Which approach is used may
depend on the individual process.
However, as a general rule, this author has held the view
that the development phase is the place to clearly establish the
boundaries of the operating ranges, and how each extreme between
parameters affects the final outcome of the process. Process
validation, by definition, is used to demonstrate consistency and,
thus, should target the desired operating parameter. From a
manufacturing standpoint, it is advantageous to operate based upon
the targeted parameter values for batch consistency.
The
Process Development Report
The
Process Development Report (PDR) is the starting point for any
process validation activity. The PDR should compile reports and data
on the process starting from the first laboratory preparation and
continuing through pilot scale on to commercial production. Ideally,
interim reports should be prepared during the development process,
that will make the compilation of the PDR easier to
manage.
The PDR
should be able to tell the story of how a process evolved from the
first laboratory scale synthesis to the applicable scaled up
version. The PDR should also provide data and references that
supports the current process, identify the critical process
parameters and how to control them, and describe the expected
impurity profile. The PDR should describe the characterization of
the major impurities that compose the impurity profile, and the
development efforts to minimize or eliminate impurities.
Although the PDR is a technical document, the data contained
therein supports the validation activity. The PDR should be used as
a basis for developing the validation parameters for the product in
question. The PDR should summarize the data in a level of detail
that is sufficient to describe the development cycle for the
process. Any reader should be able to get a thorough understanding
of the process history by reading the report.
Development
reports can be (and frequently are) requested during a regulatory
inspection. Although QA may not be required to approve a development
report since it is a technical report (although it is highly
recommended), QA should audit the development report for complete
traceability to the raw data. This is particularly recommended for
data that either supports a process step as critical or not.
Ideally, this is done prior to approving process validation
protocols.
The PDR
is a living document and should be continuously updated through the
product lifecycle. Many firms create a PDR and then ignore it once
process validation starts or is completed. Since improvements to a
process can be made throughout the lifetime of the process, these
improvements may require additional validation work and regulatory
filings. These changes and improvements should initiate an update of
the PDR. At a minimum, such documentation practices reflect good
scientific discipline.
The
Process Validation Master Plan
The
process Validation Master Plan (VMP) is the blueprint to all of the
process validation activities. The VMP should describe the entire
synthetic route to the API, identify which process steps require
process validation, identify starting and raw materials and the
sources of these materials. If process validation will be performed
at any contact manufacturer that is used to provide an intermediate
or API, the VMP should describe the relative responsibilities for
each organization.
Figure
3 lists some of the components that should be included in a VMP. The
VMP should not be too detailed as the validation protocol itself is
really the document that should provide all of the details. The VMP
is also a living document, and should be amended or updated as
needed if the plan changes, with the appropriately documented change
justification. However, good thought and planning should be put into
the original VMP since too many changes to a VMP may give an
impression of a poorly planned validation or weak validation
program.
The VMP should be readily available to all personnel
who will be critical players in the validation exercise, including
contract sites, as applicable.
The
Process Validation Protocol
The
process validation protocol will describe, in detail, the “how to”
for the validation exercise. Any procedure should be either
described in detail in the protocol or a reference should be
provided to an already established procedure for handling the task.
The protocol should be used in training all individuals who will be
involved in process validation.
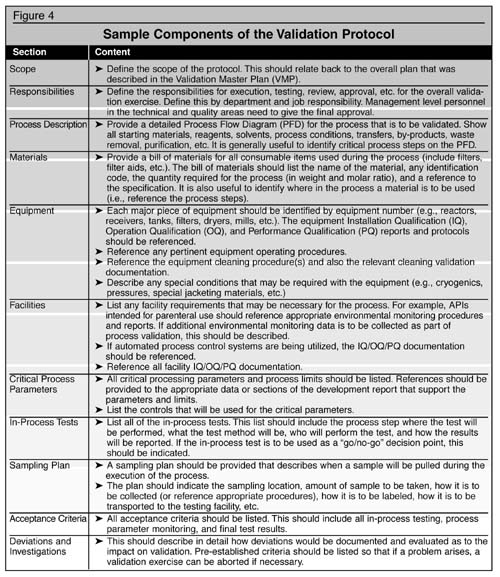
There
are many components to the protocol and the more critical components
are described in Figure 4. All components of the validation should
be described in the protocol. These would include equipment and
facility qualification documents, materials and specifications,
sampling procedures, analytical testing (both in-process and final),
analytical methods, etc.
The
protocol should be drafted to be as concise and instructive as
possible. Where applicable, data should be entered directly into the
protocol. If an exercise is particularly complex, a series of
protocols may be preferable. Protocols should have places for
reviewers’ initials or signatures.
Essentially, the protocol resembles a master record in many
ways, but it does provide a greater level of descriptive detail
regarding the execution of the process, monitoring of the critical
steps, sampling instructions, etc. It is not necessary to repeat all
of the instructions that are in the master record, however, critical
steps and operations that are unique to the exercise should be fully
described.
Critical Parameters
The
identification of critical process parameters is the key to a
successful process validation exercise, and to maintaining process
consistency throughout the process lifetime. The processing
parameters need to be evaluated carefully throughout the development
process to truly determine the critical parameters (this is where a
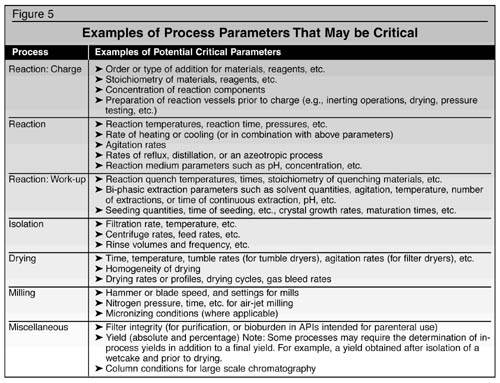
well-written PDR is most useful). Figure 5 provides some examples of
parameters that may be determined to be critical in a chemical
process. A critical parameter can represent a chemical reaction
issue, an engineering issue, or a combination of both.
In some
cases, process parameters may need to be classified in different
categories than either critical or non-critical. API manufacturing
can be very complex, and a parameter may be considered critical, but
not necessarily lethal to the process. One challenge to any process
validation program is to clearly define the various types of
critical parameters. The following is an example of how this might
be approached:
Process Critical Parameter: A process parameter that
if not maintained within established limits would lead to a process
failure. This would require routine in-process testing to ensure
maintenance of the parameter. For example, a reaction is run at 50
±2ј C, but the product will rapidly decompose starting at 58ј C.
This temperature is critical, and if not maintained, could lead to a
failure.
Process Value Parameter: A process parameter that if
not maintained within the established limits could impact the normal
course of the process (but may not lead to a failure). This type of
parameter may require strict monitoring and verification, but may
not require routine in-process testing to control the outcome. For
example, an ionic aqueous solution is required for extraction of an
organic phase. If the ionic strength is insufficient, an emulsion
can form, leading to longer settling times or indeterminate phase
separation. In this case, the length of the process could be
affected without having impact on the overall batch.
The key
to defining critical parameters is in understanding all aspects of a
given process, and ensuring that the right level of control is given
to the parameters that need it. One common mistake that is made is
defining critical parameters only in terms of the chemical process;
engineering parameters frequently are overlooked. It is important to
reinforce the concept that a chemical process is the intertwining of
both chemical and engineering aspects.
Some
organizations chose to define parameters only if they could affect
the impurity profile or the ability of a material to pass
specification. However, the ability of a material to meet
specification is not the sole indicator that the process is running
smoothly or that it is validated. Critical parameters can also
affect things such as yield, without affecting the material
specifications. Therefore, the validation protocol should establish
criteria for yields, and should be consistent with normal process
yield experience during development. Abnormal yields, such as too
high or too low, should be a red flag that there may be a problem. A
high yield, for example, could mean an unexpected contamination with
some salts, incomplete or non-uniform drying, or some other serious
problem that could impact the quality of the final
product.
Finally, the degree to which a critical parameter can be
monitored is dependent on the choice and sensitivity of the
analytical method. Therefore, it is important to reiterate that the
analytical method development and validation program has equal
importance towards the success of process validation.
Equipment and Facility Qualification
In
general, it is best to have all facility and equipment
qualifications completed prior to process validation (certainly all
IQ/OQ should be completed). Performance Qualification (PQ) can be
performed during process validation, but it is generally advisable
to have completed this testing prior to process validation since it
would add additional risk to the exercise. However, there may be
some instances where it is useful to concurrently perform an
equipment or facility PQ during process validation. In those cases,
a separate PQ protocol should be drafted and referenced in the
process validation protocol.
The
Process Validation Report
The
process validation report should summarize the results of the
process validation exercise. If a multi-step synthesis is being
validated, each process step that has a validation protocol
associated with it should have a final report for that step. A final
governing validation report should be written that summarizes the
entire effort. A report should be written even if a problem was
encountered during the execution of the validation and it was
unsuccessful. In this case, the report should describe the
investigation conclusion, and the anticipated course of action in
correcting the problem.
The
validation report is also a scientific document, and as such, should
be concise and technically clear. The results of the validation
exercise should be summarized (all raw data should be referenced for
complete traceability), preferably in tabular form with the
acceptance criteria (as stated in the protocols) tabulated as well.
The acceptability of the results versus the pre-defined acceptance
criteria should be indicated by either a passing or failing
notation. In cases where a criterion failed, a discussion should be
given concerning the failure, and include the cause of the failure,
the affect of the failure on the validation, corrective actions,
etc. All deviations should be listed, and a brief summary of each
given along with an impact assessment. A definitive statement should
be made as to whether all of the criteria have been met and the
process is considered validated. Failure to provide a definitive
conclusion is an error that is seen commonly in validation reports.
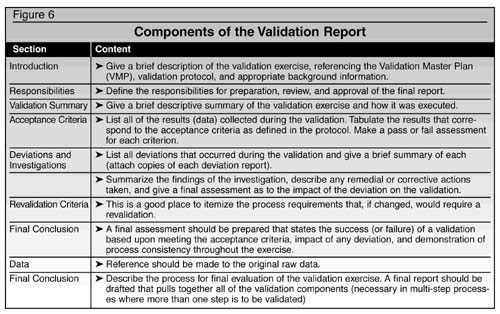
Figure 6 outlines some sections and content of a validation summary
report.
The
report should also identify the process parameters and components
that, if changed, would require additional validation (i.e.,
revalidation). Reference should be made to the current change
control procedures that will govern those changes.
Process Validation Deviations and Failures
Clearly, it is foolhardy to begin process validation without
an assurance that the process is under control, and this is related
to the process development program in general. However, even the
most well developed process will encounter a deviation or problem.
How this problem is handled can make the difference between a
successful process validation or validation failure. One common
theme when reviewing FDA Warning Letters concerning process
validation is what companies do (or don’t do) when validation
problems are encountered, even if the problem is major and the
answer is clear (e.g., recognizing a validation failure). In many of
these cases, it becomes clear that the process was not ready for
validation to begin with, and a price is now being exacted for
rushing or cutting corners. One common cause of these failures is
due to not properly identifying the critical process parameters.
Most
scientists will agree that it is more cost effective to spend time
up-front to properly develop a process, rather than attempting to
fix it after problems are encountered. Nevertheless, many companies
insist on doing the opposite by attempting to validate a process
that is not fully developed or in control. Of course, there are
constant budgetary pressures that apply to any development program.
However, with proper planning and a good, proactive program, it will
provide a better tool to project the appropriate budgetary needs,
and develop a good process that will pay for itself in the end by
being prepared for validation.
Major
problems are generally obvious in the outcome. Batch failures that
cannot be attributed to operator error or equipment breakdown
unrelated to maintenance issues are considered validation
failures.1,2 What about problems that are less clear?
The key is a thorough and well-documented investigation. In the end,
a decision must be made regarding the impact of the deviation or
problem on the validation exercise. This is also where a clear set
of pre-defined scenarios (in the protocol or VMP) can make the path
very clear. For example, if a failure is encountered due to (blank),
then do (blank). The advantage of pre-defined scenarios has already
been discussed, but this also includes management approval that
there are occurrences and conditions which everyone agrees in
advance will require a fresh start.
The
approach to an investigation is the same for any deviation, and goes
back to a fundamental approach to scientific inquiry. The following
steps should be performed as soon as possible after the problem is
discovered:
- Document clearly what occurred and when it occurred.
- Interview personnel as soon as possible (before memories
can get clouded) and document the interview.
- Collect any additional data (as is relevant).
- Describe a sequence of events of when and what happened,
review batch records, and corroborate events through data and
records.
- Document any and all remedial actions that may have been
taken.
- List
all possible causes for the event.
- After investigating, eliminate causes that do not fit the
data.
- Establish a cause or most probable cause based upon the
data.
- List
corrective actions that need to be implemented.
Both
from a regulatory perspective and a scientific perspective, any
problem that was related to the process means that the validation
was impacted. If the problem was serious enough to cause a failure,
the validation was not successful. Only events that are not
process-related could be written off as not negatively affecting the
validation (e.g., power failures, natural disasters, human error,
etc.). Even if a process validation failed and the cause was not
process-related, the validation should be replaced with a fresh
run.1 Ideally, an interim summary report can close the
event, and the VMP should then be amended to include the additional
process validation run(s). In all cases, the QA unit is required to
approve the documented investigation, conclusions, remedial and
corrective actions, assessments of impact, etc.
Process Validation, Third-Party Manufacturing, and the
Virtual Company
In a
contract situation, all process validation responsibilities should
be fully described in a quality agreement between the two parties.
The contractee, who is typically the owner of the Investigational
New Drug (IND), New Drug Application (NDA), or Abbreviated New Drug
Application (ANDA), is ultimately responsible for the product
supplied from the contractor. This means that you must be a part of
reviewing and approving validation documents (protocols, reports,
and data). Since the contractee is ultimately responsible for the
work of the contractor, the virtual company should prepare a
comprehensive validation master plan, even though the validation
work will be performed by a contract organization. The VMP should
fully describe the roles and responsibilities of each organization
in addition to the items previously discussed.
Establishing clear-cut policies and expectations is of
particular importance when dealing with contract organizations.
European firms typically have a different view on validation and
qualification than in the U.S. Thus, the concepts of process
validation may not be as universally interpreted as your
organization’s definition. Therefore, it is important to define
these expectations early on in the relationship (preferably during
the evaluation phase) so that no surprises are discovered when it
counts. These expectations are best defined in a good quality
agreement.4
For
contractors, it is important to recognize the value in providing a
detailed quality agreement or a written set of expectations from
your customer (not all virtual companies may be experienced enough
to bring this to the table). It is incumbent on either side to take
the lead in these situations and be preemptive to avoid any
potential misunderstanding. Once there is a problem, especially
during or after process validation, there will be a tremendous
effort required from both organizations to correct the
situation.
Finally, it is important that the virtual company have a
person-in-the-plant policy, especially during process validation.
Although you have hired another company to manufacture (perhaps even
develop the process) for you, you must share in the knowledge and
expertise of the process.
Special Topics
Homogeneity
The
homogeneous nature of the final API is a very critical
characteristic that must be demonstrated, and this should be done
during process validation. Ideally, homogeneity should be
demonstrated at that stage of the process where it is expected to be
achieved, not necessarily as a final packaged product. The process
step where homogeneity testing is frequently employed is the drying
step. Typically, three types of dryers are used:
- Tray
dryers
- Tumble dryers
- Filter dryers
In each
case, the validation protocol should include a sampling plan and
frequency to test various locations and times of the drying process.
These data should demonstrate the product uniformity during the
drying process. Sampling and testing may also be desirable on the
filtered wet cake prior to any drying activity to demonstrate
homogeneity of crystallization or filtration. Testing at this stage
can be focused on attributes that are applicable such as assay,
impurity profile, Organic Volatile Impurities (OVI), loss on drying,
moisture, etc.
Finally, the packaged product should undergo a comprehensive
homogeneity sampling and testing regimen. This should include a
combination of physical (appearance, particle size, powder flow,
etc.) and chemical tests (assay, impurities, heavy metals, OVI,
etc.). A sampling plan should be developed that provides a good
matrix for demonstrating homogeneity. The plan should take into
consideration the size and volume of the final packaged product. If
a material is easily compressed, a compression study should be
included, and may be included with a shipping study (see section
below).
Shipping Studies
The
final packaged API should be subjected to shipping studies.
Stability programs are designed to show the stability of a material
under storage, but they do not test movement of the product during
shipment. A shipping study can be performed at any time once the
final packaging and shipping characteristics are identified.
However, it is often convenient to include it in the validation
exercise. A shipping study should incorporate the use of portable
temperature and humidity monitors to show the environmental
conditions that the material has been subjected to during the normal
course of shipping. Stress tests of the packaging materials may be
performed, but should probably be performed on a suitable placebo.
Stress testing should address the conditions of shipment that could
impact the quality of the material being shipped (e.g., temperature,
humidity, permeability of primary and secondary containers,
etc.).
APIs
that have stringent storage requirements (e.g., refrigeration or
frozen), should have equally strict acceptance criteria for the
shipping study. If a special shipping container is required, or if a
consumable coolant (e.g., dry ice) is utilized, the protocol should
include all procedures for preparing the shipment, training of
personnel, etc.
Stability Testing
API:
Each process validation batch should be placed in a stability
program. Ideally, stability data has already been accumulated to
establish a retest period for the API. The stability protocol can be
either a standard protocol, or drafted with the validation protocol
specifically for the validation exercise, and should include normal
and accelerated storage conditions per the International Conference
on Harmonization (ICH) guidelines.3
Intermediates: If an intermediate is going to be stored or
stockpiled, stability data and retest periods need to be
established. Normally, it is a good idea to perform a hold study on
intermediates (e.g., one month, two month etc.) in the event of
delays in manufacturing, campaigning, etc. The hold study does not
necessarily need to be a part of process validation, but should be
performed prior to process validation and under a specific
protocol.
Monitoring and Trending
The key
to maintaining a process is through monitoring and trending the
process. An extensive database should be established that collects
process data. Typically, the critical quality attributes and the
critical process parameters are monitored; however, a particular
process may require additional factors to be monitored.
In
establishing a starting point for a process monitoring program, a
meaningful data baseline needs to be established, and should be from
a process that has the parameters of the process well defined.
Although it is always beneficial to monitor process parameters
during development, a baseline will probably not be initially
established until the first validation batches are produced.
However, if there were demonstration batches or other batches
produced prior to process validation that are equivalent to the
validation batches, these could represent a starting point. There
may be processes where even early development batches have enough
definition to begin this evaluation. However, enough batches need to
be produced to provide a sound statistical base for data evaluation.
This number probably should be evaluated on a case-by-case basis,
and be based on the process and process attributes that are being
monitored (since the confidence level is dependent on the number of
batches).
An
Annual Product Review (APR) is a quality requirement, but in fact,
this review should be performed routinely by the appropriate
technical group. Process trends should be investigated and
addressed, as necessary. This activity should also be captured in
PDR updates if there is additional process improvement work that is
performed. There are several good software packages available that
will assist with routine monitoring and data handling.
Conclusion
Successful process validation programs begin with a
thoughtful and comprehensive corporate policy concerning the process
validation program. This policy should recognize that process
validation begins at the initial stages of development, and does not
end until the lifetime of the product is over. It is important that
all employees be fully trained and understand their role in the
program. Good science, well-documented development programs,
proactive procedures and definitions, and well-written protocols
will increase the chances of successful process validation.
Finally, process validation does not end at the successful
completion of the exercise or final report. Process validation is a
lifetime event that requires continuous process monitoring,
trending, and evaluation.
About the Author
Roger
W. Koops, Ph.D., is currently the Associate Director of Quality at
Genelabs Technologies, Inc. and directs the Quality Assurance and
Quality Control groups. He has over 11 years of experience in API
process development, manufacturing, and quality related areas
including process validation, compliance evaluation of API
manufacturing, equipment and facilities, third-party manufacturing,
and quality systems. Dr. Koops received his Ph.D. degree in
Chemistry from the University of California, Riverside, and his
undergraduate degree from Western Washington University. He can be
reached by phone at 650-562-1441, by fax at 650-368-3198, or by
e-mail at rogerk@genelabs.com.
References
- FDA.
Guidance For Industry (draft): Manufacturing, Processing, or
Holding Active Pharmaceutical Ingredients. 1998.
- ICH
Q7A (draft): Good Manufacturing Guide for Active Pharmaceutical
Ingredients. 1998.
- ICH
Q1A(R): Stability Testing for New Drug Substances and Drug
Products. 1994, (R): 2000.
- Bobrowicz, G., The Quality Agreement: Compliance
Considerations in Selecting a Contract Manufacturer. BioPharm.
Feb., 2001. p 14.
Article Acronym Listing
ANDA:
Abbreviated New Drug Application
API: Active Pharmaceutical
Ingredient
APR: Annual Product Review
cGMP: Current Good
Manufacturing Practice
ICH: International Conference on
Harmonization
IND: Investigational New Drug
IQ: Installation
Qualification
NDA: New Drug Application
OQ: Operational
Qualification
OVI: Organic Volatile Impurities
PDR:
Process Development Report
PFD: Process Flow Diagram
PQ:
Performance Qualification
QA: Quality Assurance
QC: Quality
Control
SOP: Standard Operating Procedure
VMP: Validation
Master Plan |