Analytical Chemistry
May 1, 1996
Analytical Chemistry
1996, (68) 305A-309A
Copyright © 1996 by the American Chemical Society.
A Practical Guide to Analytical Method Validation
Doing a thorough method validation can be tedious, but the consequences of not doing it right are wasted time, money, and resources
The ability to provide timely, accurate, and reliable data is central to the role of analytical chemists and is especially true in the discovery, development, and manufacture of pharmaceuticals. Analytical data are used to screen potential drug candidates, aid in the development of drug syntheses, support formulation studies, monitor the stability of bulk pharmaceuticals and formulated products, and test final products for release. The quality of analytical data is a key factor in the success of a drug development program. The process of method development and validation has a direct impact on the quality of these data.
Although a thorough validation cannot rule out all potential problems, the process of method development and validation should address the most common ones. Examples of typical problems that can be minimized or avoided are synthesis impurities that coelute with the analyte peak in an HPLC assay; a particular type of column that no longer produces the separation needed because the supplier of the column has changed the manufacturing process; an assay method that is transferred to a second laboratory where they are unable to achieve the same detection limit; and a quality assurance audit of a validation report that finds no documentation on how the method was performed during the validation.
Problems increase as additional people, laboratories, and equipment are used to perform the method. When the method is used in the developer's laboratory, a small adjustment can usually be made to make the method work, but the flexibility to change it is lost once the method is transferred to other laboratories or used for official product testing. This is especially true in the pharmaceutical industry, where methods are submitted to regulatory agencies and changes may require formal approval before they can be implemented for official testing. The best way to minimize method problems is to perform adequate validation experiments during development.
What is method validation?
Method validation is the process of
proving that an analytical method is acceptable for its intended purpose. For
pharmaceutical methods, guidelines from the United States Pharmacopeia (USP)
(
Although there is general agreement about what type of studies should be
done, there is great diversity in how they are performed
(
In the early stages of drug development, it is usually not necessary to perform all of the various validation studies. Many researchers focus on specificity, linearity, accuracy, and precision studies for drugs in the preclinical through Phase II (preliminary efficacy) stages. The remaining studies are performed when the drug reaches the Phase III (efficacy) stage of development and has a higher probability of becoming a marketed product.
The process of validating a method cannot be separated from the actual development of the method conditions, because the developer will not know whether the method conditions are acceptable until validation studies are performed. The development and validation of a new analytical method may therefore be an iterative process. Results of validation studies may indicate that a change in the procedure is necessary, which may then require revalidation. During each validation study, key method parameters are determined and then used for all subsequent validation steps. To minimize repetitious studies and ensure that the validation data are generated under conditions equivalent to the final procedure, we recommend the following sequence of studies.
Establish minimum criteria
The first step in the method development
and validation cycle should be to set minimum requirements, which are
essentially acceptance specifications for the method. A complete list of
criteria should be agreed on by the developer and the end users before the
method is developed so that expectations are clear.
For example, is it critical that method precision (RSD) be „ 2%? Does the method need to be accurate to within 2% of the target concentration? Is it acceptable to have only one supplier of the HPLC column used in the analysis? During the actual studies and in the final validation report, these criteria will allow clear judgment about the acceptability of the analytical method.
Examples of minimum criteria are provided throughout this article that indicate practical ways to evaluate the acceptability of data from each validation study. The statistics generated for making comparisons are similar to what analysts will generate later in the routine use of the method and therefore can serve as a tool for evaluating later questionable data. More rigorous statistical evaluation techniques are available and should be used in some instances, but these may not allow as direct a comparison for method troubleshooting during routine use.
Demonstrate specificity
For chromatographic methods, developing a
separation involves demonstrating specificity, which is the ability of the
method to accurately measure the analyte response in the presence of all
potential sample components. The response of the analyte in test mixtures
containing the analyte and all potential sample components (placebo formulation,
synthesis intermediates, excipients, degradation products, process impurities,
etc.) is compared with the response of a solution containing only the analyte.
Other potential sample components are generated by exposing the analyte to
stress conditions sufficient to degrade it to 80-90% purity. For bulk
pharmaceuticals, stress conditions such as heat (50 |AoC), light (600 FC), acid
(0.1 N HCl), base (0.1 N NaOH), and oxidant
The resulting mixtures are then analyzed, and the analyte peak is evaluated for peak purity and resolution from the nearest eluting peak. If an alternate chromatographic column is to be allowed in the final method procedure, it should be identified during these studies. Once acceptable resolution is obtained for the analyte and potential sample components, the chromatographic parameters, such as column type, mobile-phase composition, flow rate, and detection mode, are considered set.
An example of specificity criteria for an assay method is that the analyte
peak will have baseline chromatographic resolution of at least
1.5
Demonstrate linearity
A linearity study verifies that the sample
solutions are in a concentration range where analyte response is linearly
proportional to concentration. For assay methods, this study is generally
performed by preparing standard solutions at five concentration levels, from 50
to 150% of the target analyte concentration. Five levels are required to allow
detection of curvature in the plotted data. The standards are evaluated using
the chromatographic conditions determined during the specificity studies.
Standards should be prepared and analyzed a minimum of three times. The 50 to 150% range for this study is wider than what is required by the FDA guidelines. In the final method procedure, a tighter range of three standards is generally used, such as 80, 100, and 120% of target; and in some instances, a single standard concentration is used.
Validating over a wider range provides confidence that the routine standard levels are well removed from nonlinear response concentrations, that the method covers a wide enough range to incorporate the limits of content uniformity testing, and that it allows quantitation of crude samples in support of process development. For impurity methods, linearity is determined by preparing standard solutions at five concentration levels over a range such as 0.05-2.5 wt%.
Acceptability of linearity data is often judged by examining the correlation
coefficient and
Although these are very practical ways of evaluating linearity data, they are
not true measures of linearity (
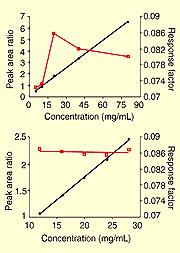
Figure 1. Peak area ratio (circles) and response factor (squares) versus concentration for mannitol.
(Top) Concentration range is 5-80 mg/mL. For peak area ration line, y=0.09775 + 0.080569x and correlatin coefficiant = 0.99952. (Bottom) Concentration range is 12-28 mg/mL. For peak area ratio line, y-0.027 + 0.08625x and correlation coefficiant = 0.99965.
If an equivalent response was obtained at each concentration, the data points would form a straight line with a zero slope. The response factors plotted in Figure 1(top) vary greatly over the range and fall only within 15% of the target concentration. A second set of mannitol data, over a narrower range of concentrations, is shown in Figure 1(bottom). The response factors for all concentrations in this range are within 1.5% of the target concentration response. The near-zero slope of the response factor plot indicates that a linear response is obtained over this concentration range.
At the completion of linearity studies, the appropriate concentration range for the standards and the injection volume should be set for all subsequent studies.
An example of a linearity criteria for an assay method is that the
correlation coefficient for each of three curves (five concentration levels
each) will be „ 0.99 for the range 80\N120\% of the target concentration. The
Demonstrate accuracy
The accuracy of a method is the closeness of
the measured value to the true value for the sample. Accuracy is usually
determined in one of four ways. First, accuracy can be assessed by analyzing a
sample of known concentration and comparing the measured value to the true
value. National Institute of Standards and Technology (NIST) reference standards
are often used; however, such a well-characterized sample is usually not
available for new drug-related analytes. The second approach is to compare test
results from the new method with results from an existing alternate method that
is known to be accurate. Again, for pharmaceutical studies, such an alternate
method is usually not available.K/p %The third and fourth approaches are based
on the recovery of known amounts of analyte spiked into sample matrix. The third
approach, which is the most widely used recovery study, is performed by spiking
analyte in blank matrices. For assay methods, spiked samples are prepared in
triplicate at three levels over a range of 50--150% of the target concentration.
If potential impurities have been isolated, they should be added to the matrix
to mimic impure samples. For impurity methods, spiked samples are prepared in
triplicate at three levels over a range that covers the expected impurity
content of the sample, such as 0.1--2.5 wt%. The analyte levels in the spiked
samples should be determined using the same quantitation procedure as will be
used in the final method procedure (i.e., same number and levels of standards,
same number of sample and standard injections, etc.). The percent recovery
should then be calculated.
The fourth approach is the technique of standard additions, which can also be used to determine recovery of spiked analyte. This approach is used if it is not possible to prepare a blank sample matrix without the presence of the analyte. This can occur, for example, with lyophilized material, in which the speciation in the lyophilized material is significantly different when the analyte is absent.
An example of an accuracy criteria for an assay method is that the mean recovery will be 100 + 2% at each concentration over the range of 80--120% of the target concentration. For an impurity method, the mean recovery will be within 0.1% absolute of the theoretical concentration or 10% relative, whichever is greater, for impurities in the range of 0.1--2.5 wt%.
Determine the range
The range of an analytical method is the
concentration interval over which acceptable accuracy, linearity, and precision
are obtained. In practice, the range is determined using data from the linearity
and accuracy studies. Assuming that acceptable linearity and accuracy (recovery)
results were obtained as described earlier, the only remaining factor to be
evaluated is precision. This precision data should be available from the
triplicate analyses of spiked samples in the accuracy study.
Figure 2 illustrates how precision may change as a function of analyte level. The %RSD values for ethanol quantitation by GC increased significantly as the concentration decreased from 1000 ppm to 10 ppm. Higher variability is expected as the analyte levels approach the detection limit for the method. The developer must judge at what concentration the imprecision becomes too great for the intended use of the method.
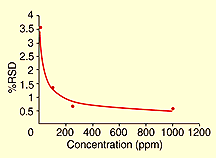
Figure 2. %RSD versus concentration for a GC headspace analysis of ethanol.
An example of range criteria for an assay method is that the acceptable range will be defined as the concentration interval over which linearity and accuracy are obtained per previously discussed criteria and that yields a precision of „ 3% RSD. For an impurity method, the acceptable range will be defined as the concentration interval over which linearity and accuracy are obtained per the above criteria, and that, in addition, yields a precision of „ 10% RSD.
Determine precision, Round 1
The precision of an analytical method
is the amount of scatter in the results obtained from multiple analyses of a
homogeneous sample. To be meaningful, the precision study must be performed
using the exact sample and standard preparation procedures that will be used in
the final method.
The first type of precision study is instrument precision or injection
repeatability (
An example of precision criteria for an assay method is that the instrument precision (RSD) will be „ 1% and the intra-assay precision will be „ 2%. For an impurity method, at the limit of quantitation, the instrument precision will be „ 5% and the intra-assay precision will be „ 10%.
Widen the scope
Once these validation studies are complete, the
method developers should be confident in the ability of the method to provide
good quantitation in
This is a good time to begin accumulating data for two or more system
suitability criteria, which are required prior to routine use of the method to
ensure that it is performing appropriately. Typically, the process involves
making five injections of a standard solution and evaluating several
chromatographic parameters (
Establish the detection limit
The detection limit of a method is
the lowest analyte concentration that produces a response detectable above the
noise level of the system, typically, three times the noise level. The detection
limit needs to be determined only for impurity methods in which chromatographic
peaks near the detection limit will be observed. The detection limit should be
estimated early in the method development-validation process and should be
repeated using the specific wording of the final procedure if any changes have
been made. It is important to test the method detection limit on different
instruments, such as those used in the different laboratories to which the
method will be transferred. An example of a detection limit criteria is that, at
the 0.05% level, an impurity will have S/N G 3.
Establish the quantitation limit
The quantitation limit is the
lowest level of analyte that can be accurately and precisely measured. This
limit is required only for impurity methods and is determined by reducing the
analyte concentration until a level is reached where the precision of the method
is unacceptable. If not determined experimentally, the quantitation limit is
often calculated as the analyte concentration that gives S/N = 10. An example of
quantitation limit criteria is that the limit will be defined as the lowest
concentration level for which an RSD „ 20% is obtained when an intra-assay
precision study is performed.
Establish stability
During the earlier validation studies, the
method developer gained some information on the stability of reagents, mobile
phases, standards, and sample solutions. For routine testing in which many
samples are prepared and analyzed each day, it is often essential that solutions
be stable enough to allow for delays such as instrument breakdowns or overnight
analyses using autosamplers. At this point, the limits of stability should be
tested. Samples and standards should be tested over at least a 48-h period, and
quantitation of components should be determined by comparison to freshly
prepared standards. If the solutions are not stable over 48 h, storage
conditions or additives should be identified that can improve stability.
An example of stability criteria for assay methods is that sample and standard solutions and the mobile phase will be stable for 48 h under defined storage conditions. Acceptable stability is „ 2% change in standard or sample response, relative to freshly prepared standards. The mobile phase is considered to have acceptable stability if aged mobile phase produces equivalent chromatography (capacity factors, resolution, or tailing factor) and assay results are within 2% of the value obtained with fresh mobile phase.
For impurity methods, the sample and standard solutions and mobile phase will be stable for 48 h under defined storage conditions. Acceptable stability is „ 20% change in standard or sample response at the limit of quantitation, relative to freshly prepared standards. The mobile phase is considered to have acceptable stability if aged mobile phase produces equivalent chromatography and if impurity results at the limit of quantitation are within 20% of the values obtained with fresh mobile phase.
Establish precision, Round 2
The remaining precision studies
comprise much of what historically has been called ruggedness. Intermediate
precision (
The last type of precision study is reproducibility
(
An example of reproducibility criteria for an assay method could be that the assay results obtained in multiple laboratories will be statistically equivalent or the mean results will be within 2% of the value obtained by the primary testing lab. For an impurity method, results obtained in multiple laboratories will be statistically equivalent or the mean results will be within 10% (relative) of the value obtained by the primary testing lab for impurities „ 1wt%, within 25% for impurities from 0.1-1.0 wt%, and within 50% for impurities \h0.1wt%.
Is it robust?
The robustness of a method is its ability to remain
unaffected by small changes in parameters such as percent organic content and pH
of the mobile phase, buffer concentration, temperature, and injection volume.
These method parameters may be evaluated one factor at a time or simultaneously
as part of a factorial experiment (
An example of robustness criteria is that the effects of the following changes in chromatographic conditions will be determined: methanol content in mobile phase adjusted by + 2%, mobile-phase pH adjusted by + 0.1 pH units, and column temperature adjusted by + 5 |AoC. If these changes are within the limits that produce acceptable chromatography, they will be incorporated in the method procedure.
Doing it right the first time
Performing a thorough method
validation can be a tedious process, but the quality of data generated with the
method is directly linked to the quality of this process. Time constraints often
do not allow for sufficient method validations. Many researchers have
experienced the consequences of invalid methods and realized that the amount of
time and resources required to solve problems discovered later exceeds what
would have been expended initially if the validation studies had been performed
properly. We hope that we have provided a guide to help you wend your way
efficiently through the method validation maze and eliminate many of the
problems common to inadequately validated analytical methods.
I wish to thank Bruce Burgess, Joseph Glajch, and the DuPont Merck radiopharmaceuticals methods quality team for their contributions in formulating many of the concepts presented in this paper.
