FDA REGULATED INDUSTRIES – VALIDATION CAPABILITIES OVERVIEW
Situation
Consumer health and safety can be put at risk if undetected problems occur during the production, storage or handling of food or pharmaceutical products. Manufacturers and government regulatory agencies have placed strict controls on the design, installation and operation of production facilities in these industries to protect the consumer.
The food and pharmaceutical industries in the USA must comply with the Current Good Manufacturing Practices (cGMPs) contained in the Code of Federal Regulations, Title 21, Food and Drugs (21CFR). The Food and Drug Administration (FDA) enforces these regulations.
Validation is one tool introduced in 21CFR that is used to assure that products are produced on a reliable and repeatable basis. Validation activities occur throughout the lifecycle of an engineering project in these industries.
Automation and information systems are a critical part of food and pharmaceutical manufacturers operations, and must be included in the validation process. TransAmerican Automation Inc. (TAI) has the capabilities to assist in this critical area. Our knowledge, experience and methodology make TAI a unique resource for these industries.
Background and Terminology
The FDA defines validation as “establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting predetermined specifications and quality attributes.” This concept applies to everything associated with making the product, including process equipment, personnel training, standard operating procedures, material handling, instrumentation, controls, and automation and information management systems (including PLCs, HMIs, DCSs, and computers). A process can only be validated once it is installed and tested in its final environment.
Manufacturers have formed various trade associations that have addressed “best practices” for validation efforts. One such organization, the Good Automated Manufacturing Process Forum (GAMP), has addressed the validation of automation and information systems in particular. In its Guide for Validation of Automated Systems in Pharmaceutical Manufacture, GAMP has expanded the concepts contained in the FDA‘s Guideline on General Principles of Process Validation. Its guide also defines the roles and responsibilities of the end user and the supplier of the automated system.
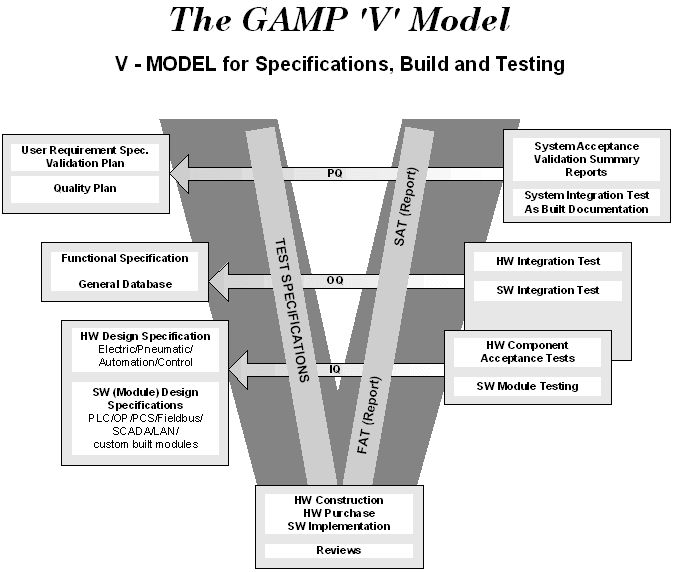
Three key components of validation are:
Installation Qualification (IQ): IQ is documented verification that all key aspects of software and hardware installation adhere to appropriate codes and approved design intentions and that the recommendations of the manufacturer have been suitably considered.
Operational Qualification (OQ): OQ is documented verification that the equipment or system operates as intended throughout representative or anticipated operating ranges.
Performance Qualification (PQ): PQ is documented verification that the process and/or the total process-related system performs as intended throughout all anticipated operating ranges. In practice, this is insuring that the system in its normal operating environment produces acceptable quality product and that sufficient documentary evidence exists to demonstrate this.
GAMP has developed a visual representation of how these three main validation components are linked back to the design process. It is called the GAMP ‘V’ Model and is shown on the next page.
Validation is a life cycle effort. Activities associated with validation will occur over the life of the installation. Ongoing process change management is a key component in maintaining the validated system.
Compliance with 21 CFR Part 11, Electronic Records; Electronic Signatures, is also a major part of the design of an automated system for a regulated industry. While not a direct validation issue, the validation efforts must assure that the operation of the final system complies with the design aspects related to 21 CFR Part 11.
TAI Services
TAI functions in the validated manufacturing process environment as either a consultant, to support the manufacturer’s technical team, or as the supplier of the automated system, or both. As a consultant, TAI can offer the services of experienced engineers to function as team members, team leaders, and/or project managers, following all applicable standards and policies.
A Validation Project Plan (VPP) is developed for each project, based on the manufacturer’s corporate policies and site specific Validation Master Plan (VMP). The VPP assigns roles and responsibilities for the various documents and tasks associated with the project. The chart on the following page illustrates how TAI’s services are blended with the manufacturer’s responsibilities for a project requiring validation.
When functioning as a system supplier, TAI follows rigorous design management practices through our internal Quality Management System (QMS), based on ISO 9000 standards. This proven methodology insures project efficiency, but with the proper attention to detail, meticulous documentation preparation, traceability, seamless hardware and software integration, and overall ease of system maintainability, essential for FDA validation compliance.
Exceptional Qualifications
TAI offers a unique blend of PEOPLE, PROCESSES and STRUCTURE, clearly focused on our Client’s needs. Our ongoing commitment to employee education, the emphasis on project management and our proven methodologies make TAI exceptionally qualified to handle the rigorous demands of process automation validation. Our services are relevant, responsive, and effective.
TAI’s operations have qualified for "Registered Member" status with the Control System Integrators Association (CSIA). TAI passed an intensive third party audit process that measured our performance against benchmarking criteria in six business areas – General Management, Human Resources, Project Management, Quality Management, Financial Management, and Business Development.
| Life Cycle Phase or Step | Client's Validation Team |
Client's Technical Team |
TAI |
| Validation Master Plan (VMP) | L/R/A | R/A | |
| Validation Project Plan (VPP) | R/A | L/R/A | P1 |
| User Requirements Specification (URS) | R/A | L | P1 |
| Audit System Supplier | R/A | L/R/A | P |
| Functional Requirements Specification (FRS) | R/A | P/R/A | L |
| Hardware Design Specification (HDS) | R/A | P/R/A | L |
| Software Design Specification (SDS) | R/A | P/R/A | L |
| Hardware Design & Implementation | R/A | L | |
| Factory Acceptance Test (FAT) Plan | R/A | R/A | L |
| Software Design & Implementation | R/A | L | |
| Installation Qualification Document (IQ) | R/A | P/R/A | L |
| Operational Qualification Document (OQ) | R/A | P/R/A | L |
| Performance Qualification Document (PQ) | R/A | L/R/A | P |
| Traceability Matrix | R/A | R/A | L |
| Factory Acceptance Testing (FAT) | P/A | L | |
| Installation Qualification Testing (IQ) | L | P | |
| Operational Qualification Testing (OQ) | L | P | |
| Performance Qualification Testing (PQ) | L | P | |
| Validation Summary Report | R/A | L/R/A | P |
