Change control
Here you will find answers to the following questions:
|
19.C.1 Principles of change control
As a rule, before a medicinal product can be manufactured, the theoretical model of the medicinal product must be specified in the form of an approval. Requirements regarding quality, efficacy and safety are thereby defined.
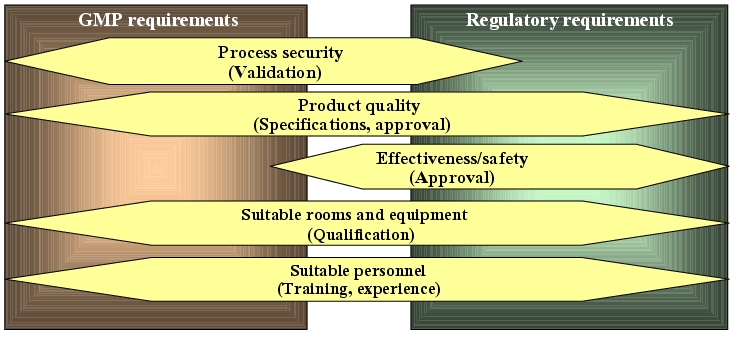
Reproducing the model in practice (GMP-conform manufacture/testing) also requires the specification of numerous requirements. The manufacture and quality control procedure should be documented in the form of instructions. The materials to be used here must be specified. The basic conditions required for a reproducible quality, such as suitable rooms, qualified facilities, trained personnel and the type of documentation, must be defined (cf. figure 19.C-1).
|

Before the requirements mentioned above can be implemented in practice, their suitability for the intended purpose must be reviewed. In the theoretical approval model, this review is carried out by the regulatory authorities as part of the authorisation procedure. The suitability and authorisation for use in practice is confirmed by a notice of approval to the applicant. In the pharmaceutical manufacturing company, the suitability of apparatus/facilities and procedures must be proven with qualification/validation. In these cases, suitability and authorisation for use in practice is confirmed with a signature on the qualification/validation report of the person responsible.
The principle that suitable requirements are available and must be adhered to, is not only valid the first time a medicinal product is manufactured or the first time a facility is used/a procedure comes into effect. This applies to the whole history of a medicinal product, a procedure or a facility and is to be guaranteed in every respect throughout.
Just like the entire batch history must be documented, requirements must also be stipulated in documents. These may be, for example, written specifications for materials or directions for procedures.
As each requirement is expressed in a corresponding document, it is clear that each change control for the requirements must also always involve a documentation control.
As a result of scientific/technical development, changes to the legal basic conditions or business restraints, requirements must be redefined, modified, enhanced or cancelled again and again in practice. In turn, this change to previously approved requirements requires a review and authorisation procedure to keep the system in its original state of proven suitability. This is the task of the change control.
Change control programs have become recognised as essential elements of the pharmaceutical quality assurance systems. In the glossary to Annex 15 of the EU GMP Guideline, there is a definition of the term "change control":
"A formal system by which qualified representatives of appropriate disciplines reviewproposed or actual changes that might affect the validated status of facilities, systems,equipment or processes. The intent is to determine the need for action that would en-sure and document that the system is maintained in a validated state." |
However, the EU GMP Guideline basically contains only a few notes about the handling of changes. Chapter 5.23 claims:
"5.23 Significant amendments to the manufacturing process, including any change in equipment or materials, which may affect product quality and/or the reproducibility of the process should be validated." |
There are also only two brief items of information in the American Code of Federal Regulations (CFR) on the topic of "change control":
§ 211.100 Written procedures; deviations. (a) "There shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess. Such procedures shall include all requirements in this subpart. These written procedures, including any changes, shall be drafted, reviewed, and approved by the appropriate organizational units and reviewed and approved by the quality control unit." § 211.160 General requirements. (a) "The establishment of any specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms required by this subpart, including any change in such specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms, shall be drafted by the appropriate organizational unit and reviewed and approved by the quality control unit. The requirements in this subpart shall be followed and shall be documented at the time of performance. Any deviation from the written specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms shall be recorded and justified." |
While in the USA the responsibility for the verification and authorisation of changes is the task of the quality control unit, in the relevant EU regulations, the responsibility is not assigned. However, as the change control is considered an essential element of the pharmaceutical quality assurance system, it is only logical to transfer the responsibility for the functioning of the change control program to the person responsible for quality assurance (QA representative, QA head).
Change control is not department-specific, rather the task of the whole company. This is due to the wide area of application of change control, as described in both Annex 15 and in the PIC/S document PI 006-2.
„Written procedures should be in place to describe the actions to be taken if a change is proposed to a starting material, product component, process equipment, process environment (or site), method of production or testing or any other change that may affect product quality or reproducibility of the process. Change control procedures should ensure that sufficient supporting data are generated to demonstrate that the revised process will result in a product of the desired quality, consistent with the approved specifications." (Annex 15, no. 43) "Change control is an important element in any Quality Assurance system. Written procedures should be in place to describe the actions to be taken if a change is proposed to a product component, process equipment, process environment (or site), method of production or testing or any other change that may affect product quality or support system operation." (PIC/S document PI 006, section 6.7.1) |
In this way, the change control monitors all types of changes which can influence the process reliability or product quality, evaluates them in reference to the relevant established requirements and determines the measures necessary for implementing the change or decides that a change should not be implemented. The change control therefore ensures that a system remains in its suitable state.
19.C.2 Introduction and operation of change control programs
"Commitment of the company to control change to premises, supporting utilities, materials, equipment and processes used in the manufacture of medicinal products is essential to ensure a continued validation status of the systems concerned. This commitment should be stated in the relevant company documentation. For example, the Quality Manual, Quality Policy Documents or the Validation Master Plan. As part of its Quality Management System the company should have a defined and formalised Change Control Procedure. " (PIC/S document PI 006, section 2.6)
The introduction of a change control program is a managerial task which must be supported by top management levels of a company. A corresponding statement for quality management can therefore be expected (cf. figure 19.C-2).
|

Many types of changes affect several regulation areas simultaneously (e.g. GMP requirements, authorisation requirements, employment protection requirements). Because quality-relevant changes can affect several areas of a company (e.g. research/development, regulatory affairs, manufacture, quality control, engineering, marketing), the control must be a task for the whole company (cf. figure 19.C-3).
|
As a rule, it is therefore not useful to operate different parallel and independently-working inter-department change control programs. In the past, the local processing of changes was carried out as the change control was not then yet recognised to be a quality assurance task for the company as a whole. Some companies are still making the mistake and work with parallel change control programs: a change control program for the EDP area, a change control program for the regulatory affairs department and a change control program for the engineering department. The collaboration of different departments in terms of change control is for this reason not always guaranteed. It has therefore been the case in practice that control software was changed in the manufacturing area, without the head of production being aware of this because he was not a member of the change control committee for the EDP department which implemented this change autonomously.
Independent change control programs cause an unwanted attitude in the department, expressed as delimitations of responsibility, ineffective inter-department communication and unfavourable procedure- and target-oriented attitudes. In addition to this, it is often the case that decision procedures and documentation formats/contents are not consistent and not well suited. This must be observed during the introduction of a change control program.
The central processing of change procedures, coordinated by the quality assurance department, has several advantages compared with local procedures:
- There is a common understanding of what a change represents.
- The classification schemes used by the persons involved to grade changes are congruent.
- The risks connected with the change can be evaluated in a multi-disciplinary manner.
- There is a uniform documentation and authorisation procedure.
The main requirement for the introduction of a change control program is a high quality awareness and an understanding for the functionality of quality assurance systems among the staff. Change control can therefore not be assigned "from above", the significance and benefits must be communicated as part of an intensive training. Changes which require control must be first recognised as such "on site" so that the necessary procedures can be initiated. Only when the staff are motivated to continuously improve the quality of the product they manufacture will they accept the change control as a suitable tool and not reject it as bureaucratic formality.
The type and scope of a change control program must comply with the individual company requirements:
- In classical GMP-relevant areas, the change control serves to maintain the validated and specified status. Validated processes and qualified facilities can be influenced in their state by changes so that the medicinal products manufactured no longer certainly comply with specifications. The same applies for changes to material specifications. A complete revalidation/requalification can then be necessary. In these cases, a formalised procedure is required to evaluate the risks associated with the change, determine the measures necessary to maintain the validation status and authorise the change after successful revalidation.
- Holders of marketing authorisations must guarantee that for changes requiring reporting or agreement, the necessary regulatory prerequisites are first met. Contract manufacturers that do not have their own authorisations and resulting obligations for reporting must guarantee that the contract giver is informed of the company-internal changes that could have an influence on their application documentation. This requires that the contract giver is included in the change control program of the contract manufacturer. Conversely, it is frequently the holder of the authorisation that causes the corresponding subsequent changes with the contract manufacturer because he changed his authorisation. Change control is in this case not only a task that applies within the company, rather a task that applies across companies. This is valid in particular for multi-national companies: changes can affect the approval status in different countries or affect the utilities at different manufacturing sites.
- In areas where medicinal products are developed, processes are optimised or clinical research takes place, the strict obligations to report that are linked with the approval do not have to be fulfilled. However, persons working in this area expect their work to be comprehensible. Changes in these areas should also be evaluated and documented in accordance with a described procedure. During the approval procedure at the latest or during inspections before the approval (pre-approval-inspection), the development of a medicinal product or process must be consistently proved.
According to the area of consideration (e.g. approval conformity or validation status), it may be necessary to use different change procedures as a base. This is the way many companies deal with changes to printed packaging material (information for use, folding cartons, labels) in accordance with a special change control procedure, because these changes occur relatively frequently in practice and the process sequences can be standardised easily. In these cases, it should be observed that the sequences and the criteria used are not independent, but are carefully matched to suit and coordinate with each other.
The examples (lists, flow charts) for grading the changes, as used in many companies, can and should not replace the individual evaluation of a change, in particular the linked consideration of risk. The example in figure 19.C-4 therefore only shows one way to grade changes. Some companies use different and extensive grading categories. Others have extensive lists of possible changes and their grades. The display of change procedures contains large flow charts which are often complicated and hard to understand. These companies have previously tried to show all potential types of changes and define the required sequences in diagrams. Such a procedure is not incorrect because it shows the scope of consideration of the change control and example help in the introduction phase for evaluation and development of changes. This can however deceive one into thinking everything is regulated. After the introduction phase, most companies find that changes do not always fit into a prefabricated chart and instead experience and know-how for particular cases is required.
| Changes requiring control |
Not requiring |
||
|---|---|---|---|
| Major (major change) |
Minor (minor change) |
||
Significance |
Influences product quality or process reliability. |
Influences a unit requiring control. |
No relevance to GMP or authorisation |
Possible measures (selection) |
|
|
|
Examples |
|
|
|
Other classifications not included in figure 19.C-4 are possible. It is not decisive which and how many change classes a company has determined, but how it is guaranteed that changes requiring control are recognised as such and implemented according to a defined procedure.
See the PIC/S document PI 006 for notes on grading changes. In chapter 6.7.4. there is a list of changes that may make a revalidation necessary (cf. figure 19.C-5).
| Changes which may require a revalidation |
|---|
|
The so called "trials" cause a problem area in change control. Trials are preliminary, temporary changes which can be permanently established or revoked after a trial period. With trials, there is a risk that these intended temporary changes gradually become permanent changes, without a formal change control procedure being carried out. Regardless of how long a trial is retained and whether it is withdrawn after a trial phase or be introduced permanently, trials should be dealt with according to the same procedure as all other changes. Before the trial phase starts, an analogous change control procedure should therefore be carried out.
Deviations should not be treated as changes, not even when deviations become changes after the failure has been clarified. A deviation is an unplanned and undesirable deviation from a requirement. It does not correspond with the aim and procedure of change control and should be dealt with according to a special procedure about handling deviations.
An important function as part of the change control program is fulfilled by the change control committee (also known as: change control team, change control panel). This permanent committee generally consists of the head of quality assurance, who frequently also chairs, and the heads of manufacturing, quality control, sales, regulatory affairs and the information representative. If necessary, further departments (e.g. research/development, EDP, engineering) also become involved. The task of the committee is to evaluate changes, determine the measures required and coordinate measures for the departments affected by the change and the final authorisation.
A major problem, especially during the introduction phase of the change control system, is the issue of which changes the change control committee should deal with. It is obvious that this committee cannot deal with all changes in a company due to capacity reasons. As a matter of fact, only changes requiring control should be processed by this committee. Firstly, these are changes relevant for the regulatory status and involve reporting or authorisation procedures. Secondly, they are changes which could have an influence on the attributes of a GMP-relevant system, facility, apparatus, material/product or a procedure/process. This influence may be critical as regards the product quality/process reliability and require a revalidation/requalification or simply require the compilation/update of documentation. The change control committee is also involved in changes for which their implementation is often extensive and coordinating measures are necessary. The committee should also deal with all changes whose grade or implementation is unclear or questionable.
| Data for the effectiveness of change control programs |
|---|
|
The question of how the committee members communicate with each other is significant. Not all change control procedures urgently require the convening of a meeting, at which many important function heads must often be present in person. In cases which are easy to make a decision about, it is also worth considering traditional paper-based circulation procedures (serial or parallel), e-mail agreements or common access to Intranet-based forms.
The committee can only deal with change applications if they are actually assigned. A high involvement is transferred to the other staff in the company. If a critical change is not evaluated as such and no formalised change control procedure is introduced, this can cause severe problems with quality.
When a change control program has been introduced, the effectiveness of the system can be reviewed using data which is easy to determine (cf. figure 19.C-6). The functionality of the system can be measured by taking into account the effort and speed of change procedures, for example, or the deviations can be examined. The effectiveness of a change control program depends on the knowledge and experiences of the staff involved. The regular training about change control procedures is therefore extremely important. Documentation procedures and communication sequences should be structured as simply as possible to enable rapid implementation.
Deviations are unplanned and generally unwanted changes. They are expressed in the form of internal and external complaints, stability problems or batch recalls. If they occur, this can be an indication that the change control program has failed. It is possible that the change to a critical process parameter or material specification has been overlooked or a negative trend in the development of process data has not been noticed in time.
19.C.3 Documentation
"All changes should be formally requested, documented and accepted by representatives of Production, QC/QA, R&D, Engineering and Regulatory Affairs as appropriate. The likely impact (risk assessment) of the change on the product should be evaluated and the need for, and the extent of Re-validation discussed. The change control system should ensure that all notified or requested changes are satisfactorily investigated, documented and authorised." (PIC/S document PI 006, section 6.7.2) "All changes that may affect product quality or reproducibility of the process should be formally requested, documented and accepted. The likely impact of the change of facilities, systems and equipment on the product should be evaluated, including risk analysis. The need for, and the extent of, re-qualification and re-validation should be determined." |
Change control requires a written procedure (change control program) to regulate at least the points shown in figure 19.C-7
| Necessary regulations of a "change control" operating instruction |
|---|
|
If the change affects a manufacturing or testing process, then the qualified person must take this into consideration when releasing the batch (cf. PIC/S document PI 006, chapter 6.7.3).
All quality-relevant changes should be documented in full and so that they are comprehensible. The records can be archived in paper form or electronically. When storing documents, make sure that all facilities, raw data and other documents relevant for the change are kept accessible.
Changes requiring control are generally documented in the form of a change request, in which the applicant for the change proposes the type of grade/evaluation of the change, specifies the time frames and measures for carrying out the change and the change is authorised or declined by the change control committee. The documentation for the change procedure should prove that the change was evaluated (risk analysis) and the subsequently defined measures were implemented as predetermined. When implementing the change, several measures must often be coordinated as regards timing and contents. A clear display of the individual measures for a project plan is useful for the coordination of complex changes.
The "change control" operating instruction in the appendix for this chapter describes the procedure of change control and contains an application form for documenting the change procedure.
Summary:
|
Company Change control procedures |
Page/pages x of y |
|
|---|---|---|
| Document number: |
Version: |
valid from: [Enter date] |
File name/path: |
||
Area of application: |
||
Key words: |
||
Replaces version: from: |
||
Changes made since last version: |
||
Cross references: |
||
References: |
||
Distribution list: |
||
Compiled by: on: |
Checked by: on: |
Approved by: on: |
1. Purpose of the instruction: Internal requirements are specified in order to comply with the legal drug product provisions and the GMP Guideline, as well as to ensure that the quality of the medicinal product complies with the approval and is reproducible. These requirements cover the entire manufacturing process and the quality control for a medicinal product, including the materials and facilities/apparatus used. The purpose of this instruction is to ensure that the quality of the medicinal product, the safety of facilities/apparatus, the safety of procedures/process and conformity with the applicable application files for marketing authorisation are maintained in the event of changes to these requirements. |
||
2. Definitions/abbreviations: Deviations: unplanned and undesirable deviation from a requirement CCC: change control committee Change: planned deviation (extension, replacement, removal, addition) as part of a requirement Change control: system with which qualified representatives from corresponding departments evaluate current or planned changes in terms of their effects with regard to a specific status. The aim is to establish precautions that are necessary to prove and document compliance with the specific status. Facility: total of all apparatus linked together with a common purpose. Applicant: person who is initiating a change with a change request Apparatus: object characterised by the technical processes carried out in it. Minor change: change which fulfils the conditions of Appendix I of Regulation (EC) no. 1084/2003 or which affects the attributes of a system, facility, apparatus, material/product or procedure/process. Impairment of the product quality/process reliability is not likely. Minor changes may require notification to the regulatory or supervisory authorities. Process: set of interrelated methods and activities which convert an input into results. System: total of all facilities linked together with a common purpose. Procedure: established way of carrying out an activity. Trial: preliminary, temporary changes which are permanently established or revoked after a trial period |
||
3. Responsibilities: 3.1. Responsibilities for the established procedures 3.1.1. The legal medicinal product responsibility for proper planning, implementation and authorisation of changes is borne by the head of production, the head of quality control, the sales manager and the information representatives for their relevant area. In particular, they must ensure that:
3.1.2. The organisational processing and documentation of change control procedures is conferred to the change control committee (CCC). The CCC comprises those in the roles mentioned above and the head of regulatory affairs and the QA representative. Other heads of department or experts may be called in at the wish of one of the members. The committee has the following tasks:
3.1.3. The chairman of the CCC is the QA representative. He has the following tasks:
3.2. Responsibility for the revision of this instruction The QA representative is responsible for checking this instruction regularly to make sure it is up to date and for revising it if necessary. He must release a new version at least every two years. |
||
4. Procedure 4.1. Basic principles 4.1.1. GMP or approval-relevant changes must only be implemented if they have been previously requested in writing and authorised. 4.1.2. Trials are also subject to this procedure. 4.1.3. Deviations are not subject to this procedure, but to operating instruction [enter doc. no./version no.] "Handling deviations". 4.1.4. Changes can be requested by any staff member using the form in Appendix 1. 4.1.5. Changes of which the grade is debatable or unclear must also be requested using this procedure. |
||
4.2. Implementation of change control procedures 4.2.1. Forms for change control procedure are issued by the QA department. If a new form is issued, a change number is automatically allocated to the change procedure by the change database and is entered on the form. 4.2.2. The applicant should specify the object of the change and the significant reasons and circumstances of the change in no. 1 on the change request. He should then sign and date it. 4.2.3. The heads of area affected by the change should have the opportunity to give their opinion on the intended change, to identify any risks and to suggest necessary measures and schedules. In any case, they should be acquainted with the planned change. This is documented in no. 2 on the change request. 4.2.4. The QA representative transfers the data required to identify the procedure into the change control database. 4.2.5. The members of the CCC jointly carry out a risk analysis of the change request under no. 3.1, classify the change under no. 3.2 and give their decision to authorise or reject the change application under no. 3.3. Authorisation may be associated with a time scale. The applicant and his head of area are informed of the decision via a copy of the completed change request. 4.2.6. A decision by the change control committee in accordance with 4.2.5 may only be made if all the information and documents relevant to the decision are submitted. If necessary, the request may be returned to the applicant for completion. 4.2.7. If the change is authorised, the CCC can establish a measures plan in no. 3.4 of the change request (tasks, responsibilities, schedule). This must be completed and authorised for the change procedure to be completed. 4.2.8. Those responsible in accordance with the measures plan shall each receive a copy of section 3.4 and shall inform the QA representative when the established measures have been completed. |
||
4.2.9. The QA representative shall collect all the completed measures plans, check them for successful completion of each task and include them with the original change request. The completed change request is archived and the result and date of the completion is entered in the change database. 4.2.10. In simple cases, in which the applicant can implement the change himself without requiring any scheduling, an electronic copy of the request may be distributed to the CCC via the internal e-mail system instead of at a meeting (circulation procedure). Each CCC member then gives his written vote regarding the change request via e-mail with electronic signature. The change request and electronic voting from the CCC member are to be archived in accordance with chapter 4.3. If there is no unanimous decision, the QA representative calls a meeting about the request. 4.3. Documentation 4.3.1. The procedure in accordance with chapter 4.2 must be documented on the "change request" form in Appendix 1. 4.3.2. If necessary, documents relevant to the decision should be added to the change request. 4.3.3. The change request and documents relevant to the decision must be kept indefinitely in the "quality assurance" area of the department. 4.3.4. If necessary for capacity reasons, the paper copy of the change request and its associated documents may be replaced with an electronic archive file. 4.4. Deviation from the procedure 4.4.1. It is permissible to deviate from the regulations in chapter 4.2 only if:
4.4.2. In the cases in chapter 4.4.1, the consent of the responsible head of area or his representative should be sought before the change is implemented. 4.4.3. Once the change has been implemented, the procedure in chapter 4.2 must be followed at the earliest opportunity. |
||
5. Appendices Appendix 1: change request form |
||
Company |
Change control procedure
|
Page/pages x of y |
||
|---|---|---|---|---|
| Document number: |
Version: |
valid from: [Enter date] |
||
| Company |
Change request [Cross reference to the operating instruction on which it is based] |
Page/pages |
||
| Change no.: |
Change designation: |
|||
1. Applicant |
||||
Name ... |
Department ... |
|||
1.1. Object of the change (mark with a cross) |
||||
o Rooms o Manufacturing facilities o Quality control facilities o Media supply o Computer-assisted system o Organisation o Contract acceptor/giver o Supplier o Marketing authorisation o Documentation |
o Manufacturing procedure o Test procedure o Cleaning procedure o Other procedure o Starting materials o Packaging material o Semi-manufactured/bulk product o Finished medicinal product o Other material ... |
|||
1.2. Description of change: |
||||
|
|
|||
2. Area head Response/risk assessment of the affected areas Area: Date and signature of the person responsible: |
||||
Area: Date and signature of the person responsible: |
||||
Area: Date and signature of the person responsible: |
||||
3. Change control committee: 3.1. Additional risk assessments: 3.2. Grade of change o Major change o Minor change o No major or minor change 3.3. Decision: o The change is authorised. Time limit for the implementation: o The measures list in no. 3.4 must be observed. o The change is not authorised. Rationale: Date and signatures of the CCC members Head of Production: Head of Quality Control: Sales manager: Information representative: Head of regulatory affairs: QA representative: Other members: |
||||
3.4. Measures list
|
Responsible ... |
Time limit ... Completed on ... |
Signature ... |
|
4. QA head Completion of procedure o The procedure has been completed correctly. o The procedure has not been completed correctly. It should be abandoned and not continued. Rationale: Date and signature of the QA head . |
||||
