Stability testing
Here you will find answers to the following questions:
|
Stability tests are an important part of the development and marketing phase of a drug product or active pharmaceutical ingredient (ICH Q1A (R2), 2000; ICH Q1B, 1996; ICH Q1C, 1996; ICH Q1D, 2002; ICH Q1E, 2003, ICH Q1F, 2003; FDA CFR, 2000). The knowledge obtained is used to determine the storage conditions (temperature/moisture), the retest period for active pharmaceutical ingredients, the maximum expiration dating period (VF, shelf life) of drug products, the correct packaging to protect the product and the transport conditions. The purpose of this is to fulfil the following objective defined by ICH (see figure 14.G-1)
Objective according to ICH Q1A (R2) "The purpose of stability testing is to provide evidence on how the quality of a drug substance or drug product varies with time under the influence of a variety of environmental factors such as temperature, humidity, and light, and to establish a retest period for the drug substance or a shelf life for the drug product and recommended storage conditions." |
.
For the purposes of stability testing, a clear differentiation is made between the development and market phases.
Development phase
During development of active pharmaceutical ingredients and drug products, a wide range of stability tests is required to obtain the registration data (see Chapter 16 Research and Development). This data should demonstrate that the company has an in-depth knowledge of how the product characteristics are influenced by environmental factors and they are used as the basis for defining the recommended storage conditions and the type of protection (e. g. packaging).
Market phase
Once approval for marketing has been granted, the manufacturer has to continue stability testing to regularly check the quality of the goods produced with respect to the established influencing factors (follw-up stability testing). However, stability tests are also carried out during this phase to ensure (change-control) that the quality of the product remains unaffected by manufacturing changes (see Chapter 19.C Change control).
14.G.1 ICH guidelines for stability tests
In the past, the requirements of authorities for stability testing have varied greatly. For companies operating globally, this meant an unacceptable amount of expenditure for the registration of an active pharmaceutical ingredient or a drug product. Efforts made to harmonise these requirements were precipitated by the ICH guidelines. However, in the regions with ICH representation (USA, Europe and Japan) different amounts of time are required before a guideline passed by the ICH Steering Committee is implemented (see figure 14.G-2).
| Timing of introduction of ICH guidelines by the regions |
||||
|---|---|---|---|---|
| Guideline |
ICH |
EU (CPMP) |
USA (FDA) |
J (MHW) |
Q1A |
Dec. 1993 |
Jan. 1994 |
Sep. 1994 |
Apr. 1994 |
Q1A (R) |
Nov. 2000 |
Nov. 2000 |
Aug. 2001 |
May 2001 |
Q1A (R2) |
Feb. 2003 |
March 2003 |
Nov. 2003 |
June 2003 |
Q1B (photostability) |
Nov. 1996 |
Dec. 1996 |
May 1997 |
May 1997 |
Q1C (new dosage forms) |
Nov. 1996 |
Dec. 1996 |
May 1997 |
May 1997 |
Q1D (bracketing and matrixing designs) |
Feb. 2002 |
Feb. 2002 |
Jan. 2003 |
July 2003 |
Q1E (statistical evaluation) |
Feb. 2003 |
March 2003 |
June 2004 |
June 2003 |
Q1F (climate zones III and IV) |
Feb. 2003 |
March 2003 |
Nov. 2003 |
June 2003 |
An additional draft of a guideline from June 1998 exists for USA which encompasses all ICH guidelines (FDA, 1998).
ICH approved a range of additional stability guidelines in 2003 that focuses on the following (see figure 14.G-2):
- Statistical analysis and interpretations of data (Q1E)
- Data for registration in climate zones III and IV (Q1F)
ICH Q1F additionally stated that ICH Q1A(R) would also have to be amended again as part of a second revision to cover the new conditions for climate zones III and IV without contradiction. An additional labelling guideline (Q1H) is planned and a guideline for generic pharmaceuticals (Q1G) has been cancelled again.
A huge benefit of the harmonisation by ICH is that the varying interpretations of storage conditions, duration of testing (timing of testing) and special testing conditions (e. g. photostability) have been standardised. This has made it possible to register the product worldwide by carrying out one stability testing program.
14.G.2 Storage and storage conditions
14.G.2.1 Standard storage conditions
The world has been divided into four climate zones with countries being assigned to the relevant zones. This is largely the work of W. Grimm (Grimm, 1985) who used the mean kinetic temperature (MKT) as the basis. The four zones are described in figure 14.G-3.
| Classification according to four climatic zones |
||||
|---|---|---|---|---|
Designation |
Temperature |
Relative humidity |
mbar* |
|
I |
moderate |
21 °C |
45 % |
11.2 |
II |
subtropical |
25 °C |
60 % |
19.0 |
III |
hot (dry) |
30 °C |
35 % |
15.0 |
IV |
tropical |
30 °C |
70 % |
30.0 |
* mbar: Water saturation vapour partial pressure |
||||
The values are derived from measurements obtained in the various zones during a one-year period (USP <1151>).
The countries have been assigned to climatic zones as follows (examples):
- Climatic zone I Great Britain, Northern Europe, Canada, Russia
- Climatic zone II USA, Japan, Southern Europe (Mediterranean region)
- Climatic zone III Iran, Iraq, Sudan
- Climatic zone IV Brazil, Ghana, Indonesia, Nicaragua, Philippines
As this classification demonstrates, approximately 90 % of the global pharmaceutical market lies in the moderate or subtropical climate zones. This has been used as the basis for deriving standard storage conditions for climate zones I and II in ICH guideline Q1A
| Standard storage conditions in accordance with ICH Q1A (R2) and ICH Q1F |
||
|---|---|---|
for climate zones I and II |
Temperature |
Relative humidity |
Long-term studies |
25 °C ± 2 °C |
60 % ± 5 % |
Intermediate conditions |
30 °C ± 2 °C |
65 % ± 5 % |
Accelerated studies |
40 °C ± 2 °C |
75 % ± 5 % |
for climate zones III and IV |
||
Long-term studies |
30 °C ± 2 °C |
65 % ± 5 % |
Accelerated studies |
40 °C ± 2 °C |
75 % ± 5 % |
:
The descriptions long-term testing or real-time testing refer to conditions under which the product will remain stable for the specified period. It is up to the applicant to decide which conditions should be selected - climate zones I and II (25 °C/60 % RH) or III and IV (30 °C/65 % RH). The accelerated testing conditions are at least 15 °C higher than the long-term study conditions and are intended to increase the rate at which degradation reactions take place thus revealing quality changes at an early stage. Intermediate conditions are applied if the quality changes occurring under accelerated conditions are too great. These conditions are also known as fallback conditions.
The ICH states that "significant changes" in a drug product is defined as
- A 5 % change in assay from its initial value
- Any degradation product exceeds the set limits
- Failure to meet acceptance criteria for appearance, physical attributes or functionality tests (e. g. colour, phase separation, resuspendibility, caking, hardness, dose delivery per actuation
- Depending on the dosage form, the pH value or dissolution rate (DR) no longer satisfies the requirements.
If the applicant selects the conditions for climate zones III and IV for the long-term study, the intermediate conditions can be excluded. Furthermore, additional standard conditions apply for less stable products (see figure 14.G-5).
| Standard storage conditions for temperature-sensitive products |
||
|---|---|---|
|
Temperature |
Relative humidity |
Refrigerator storage |
5 °C ± 3 °C |
monitored |
Freezer storage |
20 °C ± 5 °C |
- |
Note: a freezer storage temperature of -15 °C ± 5 °C is specified in the FDA draft (FDA, 1998). |
||
The storage conditions proposed by the FDA for inhalation powders and aerosols are described in figure 14.G-6.
| Standard storage conditions for inhalation aerosols (according to FDA) |
||
|---|---|---|
|
Temperature |
Relative humidity |
Long-term studies |
25 °C ± 2 °C |
75 % ± 5 % |
Accelerated studies |
40 °C ± 2 °C |
75 % ± 5 % |
The term "room temperature" is frequently used to describe the standard storage temperature. It should be noted that, according to USP, this term clearly implies content that does not correspond with the ICH definition. This is dealt with in more detail in Chapter 14.G.2.8 Labelling.
The term "stress test" is also frequently incorrectly used to describe testing untder accelerated conditions. According to ICH, this term refers to specific analyses carried out on active pharmaceutical ingredients where far greater stresses are involved. This is dealt with in more detail in Chapter 14.G.2.4 Stress test.
Following removal from storage, the samples must be analysed within a specific time limit. The standard procedure previously followed to freeze the samples after removal from storage to be pooled with other samples for analysis is not permissible. The procedure is to be defined in a SOP or guideline and could be as follows:
- Completion of analysis four weeks at the latest after removal from storage (withdrawal from climate chamber)
- Report of analysis (decision) six weeks at the latest following removal from storage
- Storage of samples in accordance with recommended storage conditions (e. g. refrigeration storage) until completion of analysis
The facilities (Climate chambers) for the storage of samples must be capable of monitoring and maintaining temperature and moisture levels within the specified tolerances. This monitoring must be documented. Companies are particularly required to control access, carry out training and compile documentation. In the event of breakdown of the control system, measures must be defined in an emergency plan.
14.G.2.2 Packaging
The packaging performs a significant function as it protects the contents (active pharmaceutical ingredient or drug product). The most important aspect with regard to correct selection of primary packaging is its tightness. However, light protection, weight and handling also play a significant role. A requirement for stability testing of drug products is that it is carried out in the primary commercial packaging that will ultimately be used. Active pharmaceutical ingredients may also be stored in packaging that has a water vapour permeability which corresponds with the commercial packaging. Water vapour permeability is used for the classification of packaging - e.g. through gravimetrical determination of water absorption on silica gel in the container at 23 °C/75 % RH after 14 days (cf. USP <671>). This means that for active pharmaceutical ingredient packaging, classification may be carried out as follows (see figure 14.G-7).
| Classification of active pharmaceutical ingredient packaging according to water vapour permeability |
||
|---|---|---|
Tightness |
Classification |
Water vapour permeability/day/litres |
very leak-tight |
A |
£ 0.5 mg |
leak-tight |
B |
£ 2.0 mg |
permeable |
C |
£ 14.0 mg |
very permeable |
D |
£ 14.0 mg |
Examples:
- Bag made of 1-layer foil (LDPE foil 60 mm): C (permeable)
- Brown glass bottle with safety closure: B (leak-tight)
- Bag made of 3-layer foil
(PETP 75 mm / Alu 8 mm / PE 12 mm) : B (very leak-tight)
14.G.2.3 Sample quantities
As a general rule, sufficient quantities of samples must be stored so that any test feature can be repeated if necessary. In practise, many companies store twice as many samples as are required in case an analysis needs to be repeated from scratch. The analyses carried out and not the storage tend to be the decisive cost factor in stability studies. When reducing the scope of the analysis (Chapter 14.G.4.1 Bracketing and Chapter 14.G.4.2 Matrixing), the full design samples must also be stored as the reduction only applies for analytical testing.
14.G.2.4 Stress test
According to ICH, this includes special analyses of active pharmaceutical ingredients where stresses are increased to actively promote the degradation of an active pharmaceutical ingredient for the purposes of recording possible degradation products and understanding fundamental degradation mechanisms (known as forced decomposition test or forced degradation study). This is an important test in the first development phase for which more stringent conditions apply than the accelerated study.
| Classification of active pharmaceutical ingredients according to stability |
||
|---|---|---|
Category |
Abbreviation |
Sum of degradation products |
very stable |
S |
< LOQ* |
stable |
S |
LOQ*-1.5 % |
unstable |
I |
1.5 %-10.0 % |
very unstable |
I |
> 10 % |
* LOQ = Limit of Quantitation |
||
For example, the thermal stability is tested by increasing the temperature in 10 °C increments starting at 50 °C. The relative humidity is also 75 % or higher. In addition, oxidative, reductive and photolytic conditions are chosen in order to record substance characteristics and perform the classification accordingly. Changes in quality are also measured in aqueous solutions with varying pH values. Using the results as the basis, stability classes can be defined according to the following example (see figure 14.G-8 and figure 14.G-9).
| Classification of active pharmaceutical ingredients according to hygroscopic behaviour |
|||
|---|---|---|---|
Category |
Abbreviation |
Conditions (24 h) * |
Water absorption |
non-hygroscopic |
NH |
92 % RH |
< 0.2 % |
slightly hygroscopic |
LH |
80 % RH |
0.2 %-2.0 % |
hygroscopic |
H |
80 % RH |
2 %-15 % |
extremely hygroscopic |
SH |
80 % RH |
> 15 % |
* 80 % RH in accordance with Ph.Eur. ** if > 0.2 at 92 % RH |
|||
When carrying out stress tests, the conditions should be appropriately modified if too many degradation products are formed.
14.G.2.5 Freeze test
The aim of this test is to ensure that the quality of drug products will not be adversely affected by freezing and defrosting. A test design could look like this:
- Withdrawal from storage area
- Freezer storage, 1 week
- Defrost, 1 day at 25 °C/60 % RH
- Freezer storage, 1 week
- Defrost, 1 day at 25 °C/60 % RH
Following this, the physical characteristics are analysed and compared with an untreated sample.
Practical experience has already shown that while chemical stability generally improves at lower temperatures, the physical characteristics (e. g. solubility) are adversely affected. If a component precipitates during the freezer phase (e. g. excipient) and subsequently cannot be fully re-dissolved, the product quality has been adversely affected. The storage instructions should therefore inform the patient that the product should not be stored at temperatures e. g. "below 10 °C".
14.G.2.6 Temperature cycling test
The physical stability of products with a semi-solid formulation (creams, suspensions, lotions or similar) must be tested. During this test, the sample is alternately exposed to lower (refrigerator storage) and higher temperatures (40 °C) with changes being carried out frequently, e. g. on a daily basis over an extended observation period (1 month). Following this, a physical test involving the comparison with an untreated sample must be carried out to check in particular for possible crystallisation.
14.G.2.7 Special storage conditions for drug products
In addition to the ICH standard storage conditions, further test conditions must be determined for specific drug products. Dry environmental conditions (below 25 % RH) are critical for the quality of products in semi-permeable and permeable packaging (e. g. lotions, SVPs = Small Volume Parenterals and LVPs = Large Volume Parenterals, nasal sprays, ophthalmological products in PE ampoules or PE bottles). For this reason, the storage conditions described in figure 14.G-10 are recommended.
| Standard storage conditions for semi-permeable packaging |
||
|---|---|---|
for climate zones I and II |
Temperature |
Relative humidity |
Long-term studies |
25 °C ± 2 °C |
40 % ± 5 % |
Intermediate conditions |
30 °C ± 2 °C |
65 % ± 5 % |
Accelerated studies |
40 °C ± 2 °C |
maximum 25 % ± 5 % |
for climate zones III and IV |
||
Long-term studies |
30 °C ± 2 °C |
35 % ± 5 % |
Accelerated studies |
40 °C ± 2 °C |
maximum 25 % ± 5 % |
Once again, the applicant can choose the conditions for climate zones I and II or climate zones III and IV. If the latter is selected, the intermediate conditions can be omitted. As water vapour permeability is being measured, in this case, it is generally sufficient to test the packaging without the product and fill it with a saline solution instead of the expensive product.
If drug products are packaged in leak-tight containers (glass bottles, vials, sealed glass ampoules), the moisture inspection is not required. In these cases, the recommended storage conditions could be as described in figure 14.G-11. Generally, there is no advantage in providing separate facilities in these cases, instead, samples tend to be stored in normal climate chambers.
| Standard storage conditions for leak-tight packaging |
||
|---|---|---|
|
Temperature |
Relative humidity |
Long-term studies |
25 °C ± 2 °C |
Environment |
Intermediate conditions |
30 °C ± 2 °C |
Environment |
Accelerated studies |
40 °C ± 2 °C |
Environment |
The recommended storage conditions for drug products that are intended for storage in refrigerators are described in figure 14.G-12.
| Standard storage conditions for intended storage in refrigerators |
||
|---|---|---|
|
Temperature |
Relative humidity |
Long-term studies |
5 °C ± 3 °C |
Monitoring |
Accelerated studies |
25 °C ± 2 °C |
Environment |
If deep freezer storage is intended, the following is recommended (figure 14.G-13).
In this case, accelerated studies are not recommended. Instead a batch should be stored at higher temperatures (e. g. 5 °C ± 3 °C or 25 °C ± 2 °C) for a suitable period to allow for brief temperature fluctuations (also known as excursions) that are outside the storage conditions.
| Standard storage conditions for intended storage in freezers |
||
|---|---|---|
|
Temperature |
Relative humidity |
Long-term studies |
20 °C ± 5 °C |
- |
Note: a freezer storage temperature of -15 °C ± 5 °C is specified in the FDA draft (FDA, 1998). |
||
14.G.2.8 Labelling
The objective of stability testing is to obtain comprehensive knowledge about the properties of the active pharmaceutical ingredient or the drug product. The recommended storage conditions are derived from this and are to be made known to the patient. The following approximate classification can for example be carried out based on the storage temperature and protective measures (figure 14.G-14).
| Storage conditions and protective measures |
|
|---|---|
Category |
Description |
A |
Store below -15 °C |
B |
Store in refrigerator between +2 °C and +8 °C |
C |
Protect from frost |
D |
Do not store in refrigerator |
E |
Store between 15 °C and 25 °C |
F |
Do not store above 25 °C |
G |
Store below 30 °C |
H |
Protect from light |
I |
Protect from moisture |
J |
Store with drying agent |
K |
Store tightly closed |
The mean kinetic temperature is decisive for the classification. In the USP, the controlled room temperature is defined by analogy as a storage temperature of 25 °C whereby (brief) deviations (also known as excursions) of between 15 °C and 30 °C are permissible provided the resulting mean kinetic temperature does not exceed 25 °C. In the FDA draft (FDA, 1998), brief excursions of up 40 °C are permissible where the same limitation applies but these must be kept to an absolute minimum. Swissmedic has now requested a new storage instruction containing the following specific wording: "Store at room temperature (15 °C to 25 °C)".
14.G.3 Analyses
As a basic principle, stability tests must cover the entire shelf life. A maximum period of 5 years has been defined for this but for economic reasons (storage costs), a shorter shelf life is aimed at nowadays. It is not sufficient simply to document the start and the end of the overall period by carrying out analyses, data must be also obtained at defined intervals in between. Analyses are to be carried out every three months in the first year, every six months in the second year and annually for subsequent years. This is illustrated by the following overview:
| Test times |
||
|---|---|---|
Long-term studies |
ICH |
0, 3, 6, 9, 12, 18, 24, 36, 48, 60 months |
Accelerated studies |
ICH |
0, 3, 6 months |
|
USA |
0, 2, 4, 6 months |
|
Japan |
0, 1.5, 3, 6 months |
Note: For the submission, data obtained in the periods emphasised in bold must be presented. |
||
Following extensive discussions, the ICH concluded that at each accelerated study must include at least three test times. Four test times are required for accelerated conditions in Japan and additional results must be available after 1.5 months. For the USA, four test times are also required - at 0, 2, 4 and 6 months (FDA, 1998). As it is not possible to wait until the end of the overall test period before registration, minimum requirements exist. These specify that results obtained over a period of 12 months with long-term studies and results obtained over a period of 6 months under accelerated conditions must be presented. In figure 14.G-15, this have been emphasised in bold.
According to ICH, companies are expected to define tolerances for sample removal. For test times of at least 1 year, a tolerance of ± 1 month is standard. This means for example that "24 months" would be declared in the stability report for a sample removed from storage after 25 months. For shorter test times, appropriate reductions in the tolerances must be defined, e. g. ± 2 weeks for 3 months. In each case, the storage data must be precisely documented and the timing of the analyses must also be defined in a similar manner - see Chapter 14.G.2 Storage and storage conditions, e. g. within four weeks following removal from storage.
14.G.3.1 Test parameters
Stability tests are only meaningful if adequate selective and sensitive methods are available for the analysis of possible changes in product quality. A particular objective pursued at the development stage is also to develop and validate analytical methods using so-called stress tests - see Chapter 14.G.2.4 Stress test and Chapter 14.F Validation of analytical methods. It must be considered which test parameters (properties) will demonstrate quality changes in the product and which methods can be used to record these changes (known as stability indicating methods). A selection of possible test parameters that could be usefully applied for stability testing are listed in figure 14.G-16 for active pharmaceutical ingredients and in figure 14.G-17 for drug products.
| Test parameters for active pharmaceutical ingredients |
|
|---|---|
Appearance |
Loss on drying |
Appearance in solution (clarity) |
Melting point |
Colour of solution |
DSC (Differential Scanning Calorimetry) |
Content |
Thermogravimetry |
Purity |
Optical rotation |
IR |
Enantiomer purity |
Water content (Karl Fischer method) |
Particle size |
Residual solvents |
Powder diffraction (XRPD) |
| Test parameters for drug products |
|
|---|---|
Appearance |
Dissolution rate |
Odour |
Disintegration time |
Content |
Hardness |
Purity |
Loss on drying |
Additional analyses are recommended for specific drug products. For example, lyophilisates should be subjected to resolubility trials during stability tests. For injection solutions in ampoules, checks for particulate matter must be carried out at least at the development stage at the start and at the end of the proposed shelf life.
14.G.3.2 Reference samples
A special requirement of the FDA is the regular visual inspection of reference samples of representative batches (see Chapter 14.A.3.3 Reference samples) to check proper storage, integrity of sample material and container closures. For an oral solution for example, the reference sample would be "normal" and would have to be stored "upside down". The objective of the annual visual check could for example be to identify possible leaks in the packaging, formation of streaks or precipitation.
14.G.3.3 Consumption test
This test is carried out in order to simulate the prescribed withdrawal from multi-dosage containers (e. g. syrup, oral solution, ointments, eye drops). Withdrawal is continued according to the directions on the package insert until only 10 % of the contents remain. This is followed by an analysis and evaluation of the chemical and microbiological properties (poss. sterility). The result of an investigation of this kind is used for packaging information that states for example that eye drops must not be used ten days after they were initially opened and must be disposed of.
14.G.3.4 Compatibility test for injection solutions for infusions
A number of active pharmaceutical ingredients have a tendency to accumulate on plastic surfaces thus causing sub-strength infusions. The plastic containers used and other components must therefore be tested and another type of plastic (e. g. hard instead of soft plastic) selected, if required. Analyses must also be carried out to check for leachables (e. g. softening agents).
14.G.3.5 Analysis of compatibility of rubber stoppers and
plastic components
These compatibility tests are particularly relevant for liquid dosage forms. It is recommended (FDA, 1998) that, in addition to tests using standard extraction solutions described in the pharmacopoeias (e. g. USP <661>), more sensitive and specific tests are carried out. With oral solutions for example, the sample is stored upside down and the contents are subsequently analysed for possible contamination (e. g. with GC/MS). The visual inspection is extremely important as illustrated by rubber stoppers in the lids of oral solutions - a known source of contamination. In practise, there have been cases where, following a change to a new supplier, the new stopper released metal traces (e. g. zinc ions) that triggered a complexation reaction causing discolouration of the contents.
14.G.3.6 Photostability (ICH Q1B)
According to ICH guideline Q1A (R2), the analysis of photostability for new active pharmaceutical ingredients and drug products is necessary. The general conditions have been defined by the ICH in its own guideline (ICH Q1B). The purpose of this test is to demonstrate that the effects of light do not cause unacceptable changes in quality. Normally, the testing of one drug product batch is sufficient. The photostability test for active pharmaceutical ingredients is part of the stress test - i.e. it is carried out in the early stages of development. For the registration, the analysis is carried out for confirmation purposes as the validated analytical method can now be applied.
Light source
The light source must comply with the D65/ID65 specifications of ISO standard 10977 (ISO, 1993) whereby D65 corresponds with the international standard for daylight outdoor and ID65 corresponds with the international standard for indirect daylight indoor. Artificial daylight fluorescent lamps combining UV/Vis outputs, xenon or halogen lamps are suitable for this. Radiation below 320 nm should be eliminated using a suitable filter. Alternatively, the sample can be exposed to a cold, white fluorescent lamp and also a short-range UV lamp with a spectral range of between 320 and 400 nm.
Test conditions
When carrying out the test, the temperature should be carefully considered to allow the influence of light to be assessed independently. If required, a control sample wrapped in opaque foil (e. g. aluminium foil) may be placed in the sample room (dark control). The exposure must be at least 1200 klux · h and the energy produced by the short-range UV component must be at least 200 W · h/m2.
Active pharmaceutical ingredient
The properties of the active pharmaceutical ingredient must be taken into consideration during the trials to prevent sublimation, melting or other changes occurring during irradiation. The active pharmaceutical ingredient (solid or liquid) must be tested in a suitable container (glass or plastic vessel with translucent cover) and cooled during testing as required. It should be noted that in the (early) development stages during the stress test (Chapter 14.G.2.4 Stress test), impurities are deliberately produced by means of photo degradation whereas later the analysis of protective measures is the focus of investigations.
|
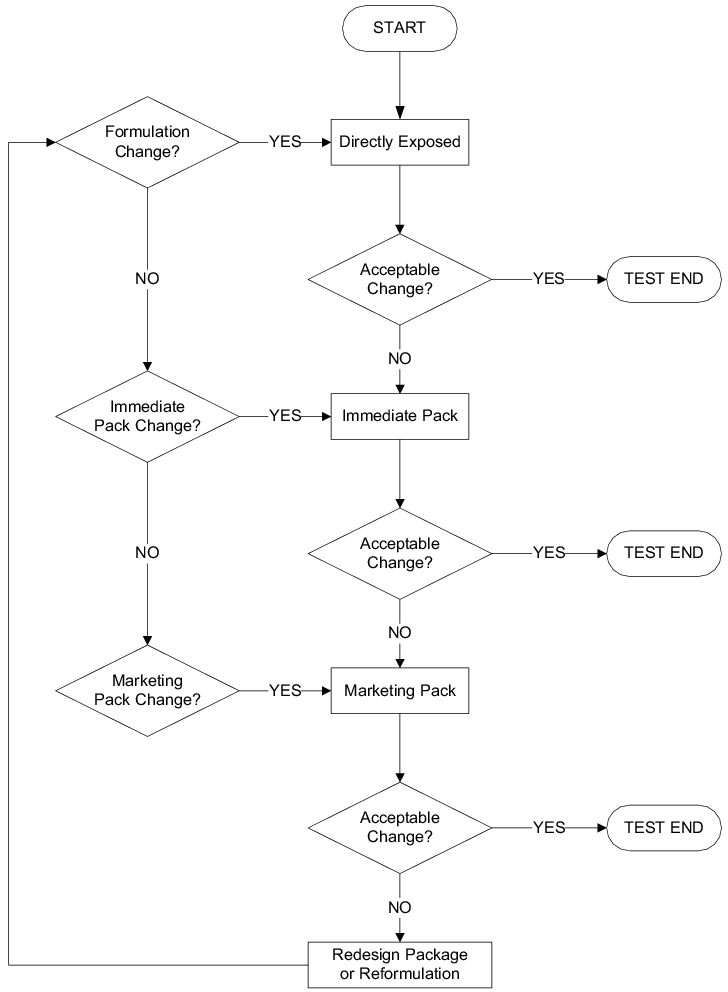
Drug product
The drug product is initially tested without packaging. If unacceptable changes occur, protection is added in stages starting with the primary packaging followed by the secondary packaging (commercial packaging). Depending on the result, the packaging must be improved (see Chapter 16 Research and Development) and/or the formulation changed. The procedure is shown in figure 14.G-18. As is the case with active pharmaceutical ingredients, the properties of the product must also be taken into account here and external influences excluded using a closed container, if required.
Analysis
Following irradiation, the physical characteristics of the samples - mainly appearance, content and purity - must be checked; in the case of drug products, e.g. also disintegration time and dissolution rate (DR). Afterwards, the necessary protective measures must be defined.
Assessment
Once the analysis is complete, the results must be assessed. The decision is made with reference to the specifications based on experience gathered during the development of the active pharmaceutical ingredient or drug product - see Chapter 16 Research and Development. The protective measures that help prevent product quality being adversely affected must subsequently be defined.
14.G.3.7 Microbiological analyses
Preservation
Sterile as well as non-sterile drug products may contain preservatives to protect the product from bacteria and fungi. The minimum content limits at the time of approval and/or end of the shelf life must be established during development. These minimum limits must be checked as part of a microbiological challenge test during which less than the required amount of preservative (e. g. ethanol) is added to the formulation in order to determine the safe range (USP <51>). The FDA recommends that these analyses are carried out at the beginning and at the end of the shelf life and every year in between, together with the assay of preservative. As already mentioned, the effects of primary packaging materials (see Chapter 14.G.3.5 Analysis of compatibility of rubber stoppers and plastic components) and normal use (Chapter 14.G.3.3 Consumption test) on the preservative must also be clarified.
Microbiological limits
During stability testing of non-sterile drug products, checks should be carried out at regular intervals to verify compliance with the microbiological limits. The USP or Ph.Eur. requirements apply in this regard.
Sterility test
The sterility of each batch of a sterile drug product should be checked at the start of the stability tests. The FDA also recommends that the integrity of primary packaging is verified annually and at the end of the shelf life. This test must be selective and sensitive enough also to detect defects caused by an insufficient barrier. The sterility test in the USP (USP <71>) is used as the basis for determining the sample size. According to the FDA, samples that have passed the integrity test may be used for other physical-chemical investigations carried out at the same test time. Under no circumstances should a sample of this kind be kept in storage and reused at a later date. The FDA also clearly states that the integrity test carried out as part of the release analysis may not be regarded as a substitute for the sterility test.
Pyrogens and endotoxins
For parenterals, the analysis of pyrogens and bacterial endotoxins should be checked at the start of and at suitable intervals during the stability tests. For most parenterals, a test at the start and the end of the shelf life will be sufficient. Products that are manufactured from dry material (powder, lyophilisate) or solutions in sealed glass ampoules also need only be checked at the start. Liquid products in plastic containers or in glass containers with a movable closure must be tested at the start and at the end as a minimum requirement. The FDA recommends that test procedures and specifications follow the FDA guideline (FDA, 1987) and USP (USP <85> and USP <151>).
14.G.3.8 Analysis of standing times
Special stability tests must be carried out to establish how long the active pharmaceutical ingredient may be stored prior to processing for formulation purposes and which protective measures must be taken. Analogous investigations must also be carried out to determine the maximum time available until the drug product bulk is packaged. The FDA accepts a maximum standing time of 30 days, data must be provided for longer periods. During the process validation (see Chapter 7 Process Validation), three batches are used to demonstrate that suitable packaging has been specified for the standing times selected.
14.G.3.9 Analysis of transport conditions
In addition to the stated storage conditions, the transport conditions must also be looked at (also see Chapter 11.M.6 Dispatch and transport). The sometimes lengthy route from the manufacturer's warehouse to the pharmacist must be monitored. It must be ensured that the drug product is intact when it finally reaches the final consumer. During these investigations, material is dispatched together with data loggers that record data on temperature and moisture. The quality of the drug product is subsequently investigated and the data obtained by the loggers is evaluated. A classification of the transport conditions is shown as an example in figure 14.G-19.
| Transport conditions |
||
|---|---|---|
Category |
Designation |
Description |
A |
Deep-freeze transportation |
Uninterrupted transportation and interim storage |
B |
Refrigerating chain |
Uninterrupted transportation and interim storage |
C |
Refrigerated transportation |
Transport and interim storage between |
D |
Controlled |
Transport and interim storage |
E |
Normal |
Transport and interim storage |
In USA, Canada and South Africa for example, transport validation is required. In these countries, not only the products themselves but also transportation by companies (carriers) is subjected to validation. The procedure could be as follows:
- The product is classified at the development phase and checks must be carried out to determine whether the product will remain intact in the packaging selected under the specified conditions.
- New products are transported three times during which the data is recorded and evaluated.
- In the next step, the carriers are evaluated to determine whether they can comply with the specified conditions (see figure 14.G-19).
- In the final stage, the carrier's compliance with the conditions is checked periodically, e. g. every two years.
14.G.4 Reduction of the study design
The scope of stability studies can be considerable, particularly towards the end of development phase and as a result of changes during the marketing phase. This is accompanied by high expenditure both in terms of personnel and finances so efforts must be made to keep the resulting costs as low as possible. The following discussion is based on the contents of ICH guideline Q1D (ICH Q1D, 2002). The aim of reducing the scope of the test is always to reduce costs and labour expenditure whilst avoiding any loss of information.
14.G.4.1 Bracketing
Bracketing is the design of a stability schedule such that only samples on the extremes of certain design factors (e. g. strength, container size and/or fill) are tested at all times as in a full design. The design assumes that the stability of any intermediate level is represented by the stability of the extremes tested.
Bracketing can be applied to studies with identical or closely related formulations, e. g. for the filling of varying capsule sizes from the same powder blend to achieve appropriate dosage strengths. Where powder blend are identical in qualitative terms but vary in terms of their quantitative composition, reasoning must be provided. Ultimately, bracketing is not intended for powder blend that vary in qualitative terms (other excipients). The following example of how bracketing may be applied is provided in ICH Q1D, 2002 (see figure 14.G-20).
| Bracketing |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Dosage |
50 mg |
75 mg |
100 mg |
|||||||
Batch |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Package size |
15 ml |
T |
T |
T |
T |
T |
T |
|||
100 ml |
||||||||||
500 ml |
T |
T |
T |
T |
T |
T |
||||
T = tested |
||||||||||
It should be noted that all samples must always be stored (full design) and bracketing must ultimately only be applied during analysis. This means that it will always be possible to subsequently analyse the remaining samples in emergencies. During planning, it is imperative that companies make provisions for the possibility that one of the extremes may be stopped during development. This would mean the loss of a primary element in the supporting structure and appropriate planning to cater for possible scenarios must be made.
14.G.4.2 Matrixing
Matrixing uses a statistical design as the basis for testing only a selected subset of factors (e. g. dosage strength, package size, filling) at a specific time point. These subsets of samples vary from one time point to the next. As a general rule, all factors must be always be tested at the beginning and the end of the shelf life. This also applies for the 12-month time point if the data of a comprehensive long-term stability study are still not available. This means that the saving in real terms (fields without "T") is not 33 % or 50 %, it is actually below this. The following examples (see figure 14.G-21 and figure 14.G-22) taken from ICH Q1D and slightly modified are used to illustrate this.
| Matrixing 1 (1/2 design, reduction by 1/2) |
|||||||
|---|---|---|---|---|---|---|---|
Dosage |
50 mg |
100 mg |
|||||
Batch |
1 |
2 |
3 |
1 |
2 |
3 |
|
Test times |
0 |
T |
T |
T |
T |
T |
T |
(Month) |
3 |
T |
T |
T |
|||
6 |
T |
T |
T |
||||
9 |
T |
T |
T |
||||
12 |
T |
T |
T |
T |
T |
T |
|
18 |
T |
T |
T |
||||
24 |
T |
T |
T |
||||
36 |
T |
T |
T |
T |
T |
T |
|
T = tested |
|||||||
| Matrixing 2 (2/3 design, reduction by 1/3) |
|||||||
|---|---|---|---|---|---|---|---|
Dosage |
50 mg |
100 mg |
|||||
Batch |
1 |
2 |
3 |
1 |
2 |
3 |
|
Test times |
0 |
T |
T |
T |
T |
T |
T |
(Month) |
3 |
T |
T |
T |
T |
||
6 |
T |
T |
T |
T |
|||
9 |
T |
T |
T |
T |
|||
12 |
T |
T |
T |
T |
T |
T |
|
18 |
T |
T |
T |
T |
|||
24 |
T |
T |
T |
T |
|||
36 |
T |
T |
T |
T |
T |
T |
|
T = tested |
|||||||
The design can also become more complex, as shown in figure 14.G-23 - figure 14.G-25 where three batches of one product with three dosages and three package sizes must be checked. This is a multidimensional design. The sample of test times is itemised in figure 14.G-23.
| Key for the test times |
||||||||
|---|---|---|---|---|---|---|---|---|
Test times (months) |
0 |
3 |
6 |
9 |
12 |
18 |
24 |
36 |
T1 |
T |
T |
T |
T |
T |
T |
T |
|
T2 |
T |
T |
T |
T |
T |
T |
||
T3 |
T |
T |
T |
T |
T |
T |
The same limitations that apply for bracketing also apply for matrixing design. The possible risks involved, particularly the low significance of the reduced quantity of data with respect to the proposed shelf life, are also explicitly stated. Due to the reduced quantity of data, it may not even be possible to provide a sound estimate of the shelf life. It is therefore urgently recommended that the design is discussed with the regulatory authorities before stability testing commences to prevent any surprises that may have an unfavourable effect on the outcome. Again, and this cannot be emphasised enough, planning must allow for the possibility that a package size or dosage may be stopped for all kinds of reasons during development thus resulting in the loss of important data.
| Matrixing test times |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Dosage |
50 mg |
75 mg |
100 mg |
|||||||
Batch |
1 |
2 |
3 |
1 |
2 |
3 |
1 |
2 |
3 |
|
Package size |
15 ml |
T1 |
T2 |
T3 |
T2 |
T3 |
T1 |
T3 |
T1 |
T2 |
100 ml |
T2 |
T3 |
T1 |
T3 |
T1 |
T2 |
T1 |
T2 |
T3 |
|
500 ml |
T3 |
T1 |
T2 |
T1 |
T2 |
T3 |
T2 |
T3 |
T1 |
|
T = tested |
||||||||||
| Matrixing test times and factors |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Dosage |
50 mg |
75 mg |
100 mg |
|||||||
Batch |
1 |
2 |
3 |
1 |
2 |
3 |
1 |
2 |
3 |
|
Package size |
15 ml |
T1 |
T3 |
T2 |
T3 |
T1 |
T2 |
|||
100 ml |
T2 |
T3 |
T1 |
T2 |
T1 |
T3 |
||||
500 ml |
T1 |
T2 |
T1 |
T3 |
T2 |
T3 |
||||
T = tested |
||||||||||
14.G.5 Stability testing in the marketing phase
14.G.5.1 Follow-up stability testing (FuST)
Once approval has been granted, the company is obliged to carry out continuous stability studies for the active pharmaceutical ingredient or drug product. The purpose of the (so-called) follow-up stability testing (FuST) is to ensure that the product quality is maintained under production conditions.
The EU has stayed abreast of this situation with the "Ad Hoc GMP Inspections Services Group" proposing the following two supplements to the EU GMP Guideline:
- Addendum to chapter 6: It is clearly stated that ongoing stability monitoring is expected.
- Addendum to chapter 1: In order to be able to prevent recalls associated with quality problems for products already introduced into the market, an overview of the product quality (process consistency and trends) must be carried out on a regular basis (product quality review).
The minimum data required (long-term testing for 12 months) is often submitted for the registration and the remaining data is then made available to the authorities at a later date. The stability reports must be updated accordingly. The testing schedule in these cases follows ICH guidelines. Additionally, at least 1 batch is to be put on stability (testing) every year. In practise, this can be carried out with relatively little effort.
The following scenario is possible:
- Storage only with storage conditions stated in the stability report (long-term study, i.e. at 5 °C or 25 °C)
- Storage in all primary packaging materials
- Storage of a sufficient quantity of samples to facilitate verification on an annual basis until the end of the shelf life
- Analysis of stability-indicating factors only
- Analysis at the "start", "middle" and "end" points of the shelf life (Note: the "start" test time includes the values for the release analysis, the "middle" test time is 1 year after the release analysis at the earliest but, as a basic principle, can be freely selected)
- Bracketing is acceptable, e. g. for 50 mg and 100 mg dosages where alternate testing of one 50 mg sample followed by one 100 mg sample is carried out
- Annual testing for USA product
This procedure is used if no change is planned for the registered parameters such as shelf life, storage conditions, i.e. only aquire data to support the "status quo". This kind of stability planning, is also sometimes referred to as GMP stability testing. The selection of any desired middle test time makes the pooling of analyses and therefore cost savings possible.
14.G.5.2 Stability commitment (SC)
The stability commitment (SC) is a special type of stability test whereby the report (SCR = Stability Commitment Report or HB = stability report, in Germany "Haltbarkeitsbericht") is specified and submitted to the authorities alongside the testing schedule. This procedure is usually applied in the following cases:
- Stability studies for active pharmaceutical ingredients and drug products following transfer to production, i.e. the first three batches manufactured once approval has been granted
- Stability studies resulting from change-control procedure (Chapter 19.C Change control), e. g. manufacturing changes, changes to the formulation or packaging, another manufacturing site
- Requests from authorities for additional data
| Application of bracketing and matrixing |
|||
|---|---|---|---|
Data material |
Product stability |
Bracketing |
Matrixing |
sufficient |
stable |
yes |
1/3 design |
sensitive |
yes |
2/3 design |
|
very sensitive |
no |
full design |
|
not sufficient |
stable |
yes |
2/3 design |
sensitive |
no |
full design |
|
very sensitive |
no |
full design |
|
In the case of the first three production batches (according to same plan as the registration batches), the authorities receive a statement about the batches that are to be stored with the storage plan and also any additional data such as the active pharmaceutical ingredient batch used, date of manufacture or similar. The batches used are normally three characteristic batches from the early phase of production.
The ICH guidelines apply for stability studies resulting from change-control procedures (see Chapter 19.C Change control). Matrixing and bracketing may be applied in this case to reduce testing. The design can be specified as shown in figure 14.G-26 (1/3 design = reduction by 2/3, 2/3 design = reduction by 1/3) depending on the existing data and stability of the product.
The authorities are informed about the results via the (SCR) report or an updated stability report (HB). Reference is to be made in the report to the registration documents (e. g. NDA number or similar). An example is provided on Page 25 that describes the Stability Commitment and includes an annex for USA.
|
|
|
|
|
|
|
|






|
|


14.G.6 Defining the retest period for an active
pharmaceutical ingredient and the shelf life for a
drug product through evaluation of stability data
(ICH Q1E)
The storage conditions and retest period or shelf life must be stated in the registration dossier for each product and each dosage in every commercial package.
The basis for this is:
- a long-term study lasting at least 12 months carried out on three batches of each dosage in the intended commercial packaging under the assumed storage conditions and
- a study lasting at least 6 months carried out on three batches of each dosage in the intended commercial packaging under accelerated or intermediate storage conditions.
Stability data are normally available for the entire proposed retest period or shelf life. The extent to which the batches vary when subjected to different storage conditions makes it possible to draw conclusions about the quality of future batches. The statistical evaluation of the data provides information whether and to what extent the retest period and shelf life can be specified by exploration beyond the period supported by existing data.
The decision tree shown in Chapter 14.G.7 Decision tree for data evaluation for retest period or for APIs or drug products (excluding frozen products) provides step-by-step instructions.
Chapter 14.G.9 Examples of the statistical evaluation of stability data provides
- an example for the evaluation of long term stability data,
- an example of a regression analysis for determining the retest period and shelf life,
- criteria for the pooling of data from different batches.
14.G.6.1 Data evaluation for the retest period for APIs and shelf life
for drug products that are intended for storage at room
temperature
1. No significant change at accelerated storage conditions
A: Long-term data and data obtained under accelerated conditions showing little or no change and little or no variability.
A statistical evaluation is not necessary for these conditions but the justification must be provided. This could include a description of the minimal changes, a mass balance and additional supporting data. Extrapolations for the retest period and shelf life may be carried out that are up to twice the length of the period supported by data but not more than 12 months beyond it.
B: Long-term data and data obtained under accelerated conditions showing changes over time and/or variability.
If changes are evident for one or several factors, a statistical analysis of long-term data is expedient in order to determine the retest period and shelf life. If stability varies between batches or other factors (e. g. content, container size), the retest period or shelf life cannot exeed the period supported by compliant data. If the changes are due to a specific factor (e. g. the dosage), specific retest periods or shelf lifes can be assigned to the various dosages. Extrapolation is also permissible, the extent of this depends on the available data:
- Data cannot be statistically evaluated or the statistical evaluation has not been carried out.
In this case, the retest period and shelf life may be extended by up to 1.5 times the length of the period supported by long-term data. The maximum permissible extension is six months. As is the case with the three primary stability batches, supporting data from registration batches with a similar formulation, smaller batch size or similar packaging are required. - Data can be statistically evaluated
If the statistical analysis is carried out, the retest period and shelf life can be extended by up to twice the length of the period supported by data. A maximum extension of 12 months is permitted. Supporting data from the registration studies are required.
2. Significant changes at accelerated storage conditions
In this case, the data for the intermediate storage conditions must be applied. Exceptions are
- changes in the hardness of suppositories that is designed to melt at 37 °C provided the melting point has been clearly demonstrated and
- failure to meet acceptance criteria for dissolution rate of 12 gelatine capsules or gel-coated compressed tablets provided the failure can be attributed unequivocally by crosslinking of the gelatine.
A: No significant changes at intermediate conditions - Data cannot be statistically evaluated or have not been evaluated
In this case, the retest period and shelf life may be extended by a maximum of three months beyond the period supported by long-term data. Supporting data for registration batches is required. - Data can be statistically evaluated
If the statistical analysis is carried out, the retest period and shelf life can be extended by up to 1.5 times the length of the period supported by data. A maximum extension of 6 months is permitted. Supporting data for registration batches is required.
B: Significant changes at intermediate conditions
If significant changes occur for one or more factors, the retest period or shelf life cannot be beyond the period supported by compliant data. In addition, a retest period or a shelf life shorter than the period supported by long-term studies may be required.
14.G.6.2 Data evaluation for period for APIs and shelf life
for drug products intended for storage
in refrigerator (2-8 °C)
The principles that apply for products stored at room temperature also apply here. Care must be taken when extrapolating values as the products are less stable. In addition, no intermediate storage conditions exist in this case.
1. No significant change at accelerated storage conditions
A: Long-term data and data obtained under accelerated conditions showing little or no change and little or no variability.
A statistical evaluation is not necessary under these conditions but the justification must be stated. This could include a description of the minimal changes, a mass balance and additional supporting data. Extrapolations for the retest period and shelf life can be made that are up to 1.5 times the length of the period supported by data but no more than six months beyond it.
B: Long-term data and data obtained under accelerated conditions showing changes over time or with respect to variability.
- Data cannot be statistically evaluated or the statistical evaluation has not been carried out
In this case, the retest period and shelf life specified may be 1.5 times the length of the period supported by long-term data. However, the maximum permitted extension is three months. Supporting data are required. - Data can be statistically evaluated
If the statistical analysis is carried out, the retest period and shelf life can be specified as up to 1.5 times the length of the period supported by data but not more than six months beyond it. Supporting data is also required in this case.
2. Significant changes at accelerated storage conditions
A: Changes occur between three and six months following storage
Long-term data is used as the basis for specifying the retest period and shelf life. If no extrapolation is possible, the shelf lifes must be shortened again depending on the availability of data. A statistical analysis can be appropriate.
B: Changes occur in the first three months of storage
Same procedure as above. In addition, the effects on stability in the event that the storage temperature is exceeded (transport, handling) should be discussed. In this case, supporting data could be obtained by carrying out a study of one batch over a maximum period of three months with accelerated conditions.
14.G.6.3 Date evaluation for retest period for APIs and shelf life
for drug products for intended storage
in a freezer (-20 °C)
The long-term data are used as the basis for specifying the retest period and shelf life. In addition, one batch should be stored at a higher temperature (e. g.: 5 °C or 25 °C) for a maximum of three months in order to obtain data that demonstrates the effect on stability of short-term excess storage temperatures (excursions).
14.G.7 Decision tree for data evaluation for retest period
or for APIs or drug products (excluding frozen products)
Figure 14.G-29 contains a graphic representation of the guidelines in the form of a decision tree (from Q1E, modified).
|

14.G.8 Procedure for statistical analysis
The purpose of this analysis is to establish, with a high degree of confidence, a retest period or shelf life during which a quantitative attribute will remain within acceptance criteria for all future batches manufactured, packaged, and stored under similar circumstances. If statistical evaluations were carried out for the registration batches, the same statistical procedure should be applied for subsequent stability commitment batches.
The regression analysis is suitable to evaluating the stability data for a quantitative attribute and to establish the retest period or shelf life. A linear correlation between the quantitative attribute and time is often assumed here. The intersection between the one-sided 95 % lower confidence level for the average value and the quantitative attribute is used as the proposed retest period or shelf life for quantitative attributes that decrease with time (e. g. content). For quantitative attributes that increase with the passing of time (e. g. degradation products), the one-sided 95 % upper confidence level of the average value is applied. The procedure can be applied for one single batch or several batches providing the data is only pooled once an appropriate statistical test has been carried out.
14.G.9 Examples of the statistical evaluation of stability data
Linear regression, poolability tests and statistical modelling can be used to evaluate quantitative results of stability analyses.
14.G.9.1 Data analysis for a single batch
Figure 14.G-30 shows the regression line for the decrease in assay of a drug product during a storage period of 48 months at 25 °C/60 % RH. The assay requirements are between 95.0 % and 105.0 %, long-term data are recorded over 12 months and the proposed shelf life is 24 months. The lower confidence level intersects the lower acceptance criterion at approximately 30 months. This evaluation therefore supports the proposed shelf life.
|

In figure 14.G-31, the situation for the main degradation product of this drug product under the same conditions is shown. The upper confidence level intersects the upper acceptance criterion at 31 months - the proposed shelf life of 24 months is therefore also supported here.
|

The final conclusion is that, with 95 % probability, the values for the assay as well as the degradation product will satisfy the acceptance criteria until the end of the newly-proposed shelf life (24 months). The usefulness of the above can only be guaranteed if the criteria described in Chapter 14.G.6 Defining the retest period for an active pharmaceutical ingredient and the shelf life for a drug product through evaluation of stability data (ICH Q1E) are satisfied. The procedure is suitable for assessing the retest period or shelf life of individual batches.
It can also be used for several batches of the same product providing the pooling of the batches has been checked using suitable statistical tests.
14.G.9.2 Data analysis of one attribute in each batch for several batches
of the same product (known as One-Factor, Full-Design Studies)
Where the classic approach to a stability test using three batches with the same dosage and packaging is used, one specific feature is initially checked to verify whether each individual batch supports the shelf life. If this is the case, no further evaluation is required and the shelf life is assured.
If the extrapolated shelf life for individual batches is below the assumed shelf life, a check can be carried out to verify whether a longer shelf life could still be specified if the data for all batches were to be pooled.
An analysis of covariance (ANCOVA) can be used to test the probability that the slopes and initial value of the regression lines (y-axis intercept) are comparable - in this case a significance level of 0.25 is assumed. If the hypothesis of the slopes is rejected, the procedure described in Chapter 14.G.9.1 Data analysis for a single batch is the only one that can be followed. The shelf life is then determined using the "unfavourable data". If the slopes are comparable, they are combined and the corresponding shelf life for each y-axis interceptor is calculated using the average value of the slopes. The shortest shelf life calculated in this manner is used as the assumed shelf life. If slopes and initial values can be combined, a joint regression line may be calculated per attribute for all batches as this generally allows a longer shelf life as opposed to separate evaluation of individual batches. The reason for this is that the confidence levels for the average value narrow down because a larger number of values are available.
It is important always firstly to check whether the slopes may be combined. The y-axis intercept should only be checked once this has been established.
Further methods are described in literature that could be useful depending on the case in hand (see literature references in ICH Guideline Q1E). If these are used, the justification as well as proof of comparability with the standard procedure (as described above) is required.
14.G.9.3 Data analysis of all attributes for several batches
(Multi-Factor, Full-Design Studies)
It is now also possible to check using a similar systematic procedure whether a longer shelf life can be assumed by combining all features of all batches. To do this, a statistical model must be created that checks the poolability of all features and combinations of features before a joint shelf life is calculated. This kind of model can also be used to evaluate bracketing or matrixing design studies. For more information, refer to the literature in ICH Guideline Q1E.
Summary Stability studies must be carried out on active pharmaceutical ingredients under stress conditions to record their degradation properties. For active pharmaceutical ingredients and drug products, data are obtained with standard storage conditions that allow the recommended storage and transport conditions, protective measures and the retest period for the active pharmaceutical ingredient or shelf life for the drug product to be determined.
|
