Sampling
Here you will find answers to the following questions:
|
Sampling is used in all areas of chemical and pharmaceutical manufacturing. It includes
- Raw materials (excipients, active ingredients, packaging materials)
- Intermediates,
- Samples taken during in-process controls,
- Finished products.
Sampling plays a central role in the entire quality control process, for the sample section taken from the batch, although only small, is considered to be representative for evaluating the entire batch quality (see figure 11). It is therefore necessary to plan and execute sampling procedures with the utmost care. Specially qualified personnel who also receive further training on a regular basis are a crucial prerequisite for this careful planning and execution.
|

It must be clear at all times - and for all the abovementioned areas of application - that the analysis of samples taken in a non-representative manner does not allow an accurate conclusion to be made in relation to the batch as a whole. Sampling errors therefore cast doubt over all the remaining stages of the quality control procedure in question. Experience has also shown that trouble shooting which is necessary as a result of the incorrect sampling is very time-consuming and problematic.
14.A.1 Requirements
14.A.1.1 Personnel
Qualities such as high observation skills and the capacity to log observations made are important when selecting personnel for sampling, in addition to personal reliability within a "first glance quality assessment procedure" (e.g. the condition of the containers from which samples are to be taken). Furthermore, special factors specified by the occupational hygiene requirements (product safety) must also be taken into consideration. Persons with infectious diseases or open wounds on uncovered parts of the body are therefore excluded from sampling activities. (see chapter 2.B Requirements of the personnel)
Personnel must be familiarised with the sampling plans and process and given further intensive training in both specialised and personal areas. They must master the techniques and the equipment and must be aware of both the risks related to sampling (e.g. contamination) and the safety measures to be followed (see figure 12)
| Training materials for sampling personnel |
|---|
|
.
14.A.1.2 Equipment
The equipment must be suitable for the intended use. It should have an inert surface and be easy to clean and sterilise after use, unless intended for single use. It must be cleaned immediately after use. The equipment must be kept clean and dry at all times and also sterilised for sterile products. If the equipment has been damaged, the damage must be clearly identified and removed. Typical excipients are dip samplers, zone samplers, pipettes, poly spoons, identification labels, control seals. There are companies who specialise in the supply of sampling equipment to the pharmaceutical industry.
14.A.1.3 Containers
The requirements relating to containers for samples can be summarised as follows:
The containers must
- be easy to fill
- be easy to empty
- not allow any raw material to be adsorbed on to the container surface
- be easy to store
- be light
- offer protection from light if required
14.A.1.4 Premises
The premises in which the sampling is carried out must not emit any particles. They must be easy and efficient to clean and sanitize if required. The indoor surfaces (ceilings, walls and floors) in particular should be kept as smooth and free of cracks as possible. Special rooms or clearly designated areas (also in the production areas) are the most suitable for this purpose. Only authorised personnel may have access when sampling is in progress. Good lighting, comfortable temperatures and humidity levels and sufficient ventilation (e.g. with a suction unit, HEPA filter, laminar flow units) must be provided in these rooms. There must be cleaning instructions in which the type and frequency of the cleaning carried out (after each sampling) must be described.
14.A.2 Sampling plan (instructions)
The sampling plan is a predefined procedure according to which one or more sub-quantities (a sample) must be taken from a batch. This includes the taking of combined samples, the subdivision of these sub-quantities into various containers and the storing of retention samples. The sample must be representative of the total quantity (random sample) (see figure 13)
| The sampling plan must clearly define |
|---|
|
.
Resampling is only possible in exceptional cases, e.g. if the sample was incorrectly prepared, was spilt or was incorrectly stored.Note: To exclude the possibility of a laboratory error following an out-of-spec result (see chapter 14.H Out-of-specification results), the same sample in question must be analysed. The size of the sample should therefore be sufficient for two chemical analyses to be carried out.
Chapter 5.30 and Annex 8 of the GMP Guideline stipulate that the identity of a batch of starting material is only assured if samples are taken from all containers and analysed (container-based identity). It is, however, permissible to take samples from just a section of the containers instead (reduced identity checking), if it is established through a validated process that the possibility of even just one container in an entire delivery being incorrectly identified can be ruled out. A supplier audit is normally a prerequisite for reduced identity checking. This audit should clarify the items listed in figure 14.
| Prerequisites which must be satisfied prior to reduced identity testing |
|---|
|
It is generally assumed that a validated process can only be accepted if the starting materials come from a manufacturer producing this product only or producing it at one company site only (monocompany). If starting materials are delivered in a sealed container or by the manufacturer directly, the manufacturer's reliability must be confirmed and their quality assurance system inspected by the buyer (pharmaceutical manufacturer) or by the competent authority on a regular basis.
It is, however, very unlikely that a procedure can be considered sufficiently validated if the delivery is carried out by a vendor from an unknown or unqualifiable source.
For starting materials for use in parenterals, a container-based identity test must generally be carried out for active ingredients. For excipients, a more restrictive approach must be adopted in relation to reduced identity testing.
The number of samples to be taken must be determined statistically in order to ensure that the number of samples is representative (see figure 15).
| Examples for number of samples to be taken |
|---|
A. Samples taken from each container for a maximum of 25 containers, samples taken from 50% of the containers for between 26 and 50 containers, samples should be taken from at least 25% of the containers if there are more than 50 containers. B. The number of containers used for samples shall equal C. 5 containers D. 1 sample per section (pallet) of a bulk delivery E. Several samples from a batch with documentation showing number and position for checking the content uniformity, no combined samples F. Further sampling plans can be authorised if required, as long as they are accurately justified. |
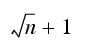
Samples for in-process controls must be taken at the beginning, in the middle and at the end of the process at least (see chapter 13.A.5 Packaging material testing).
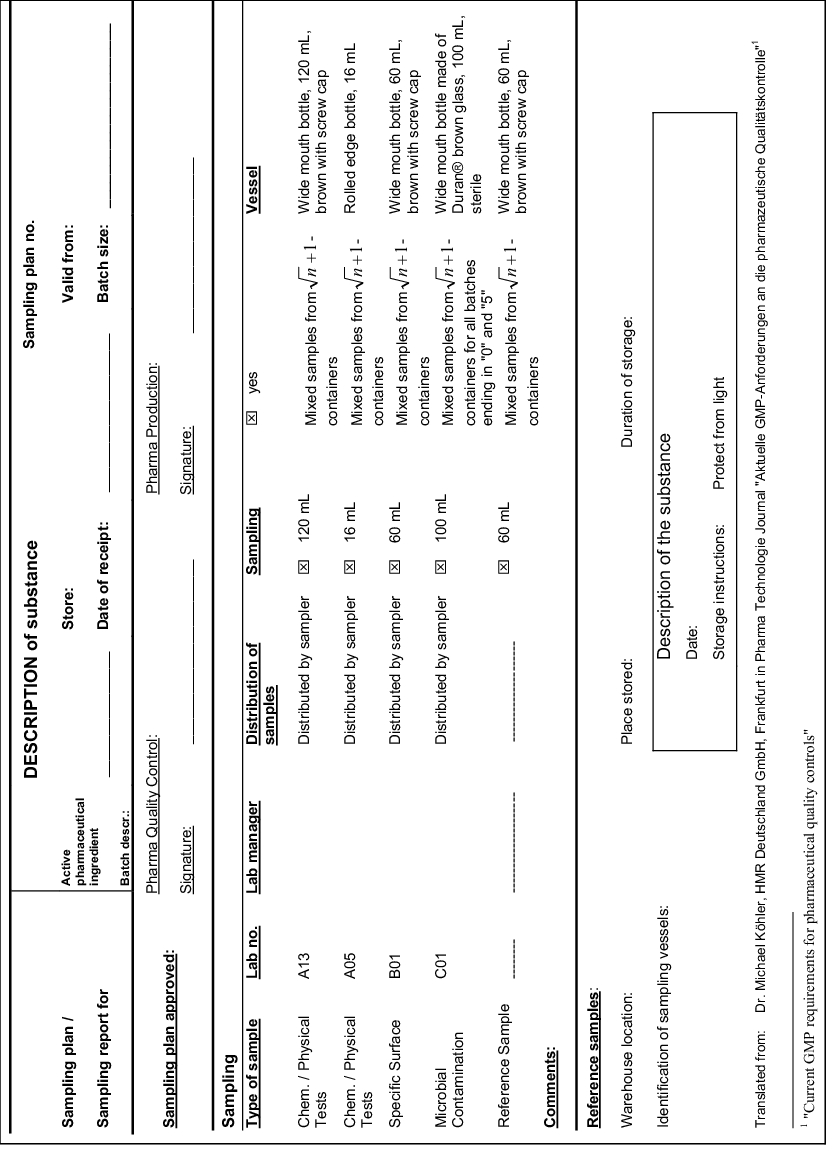
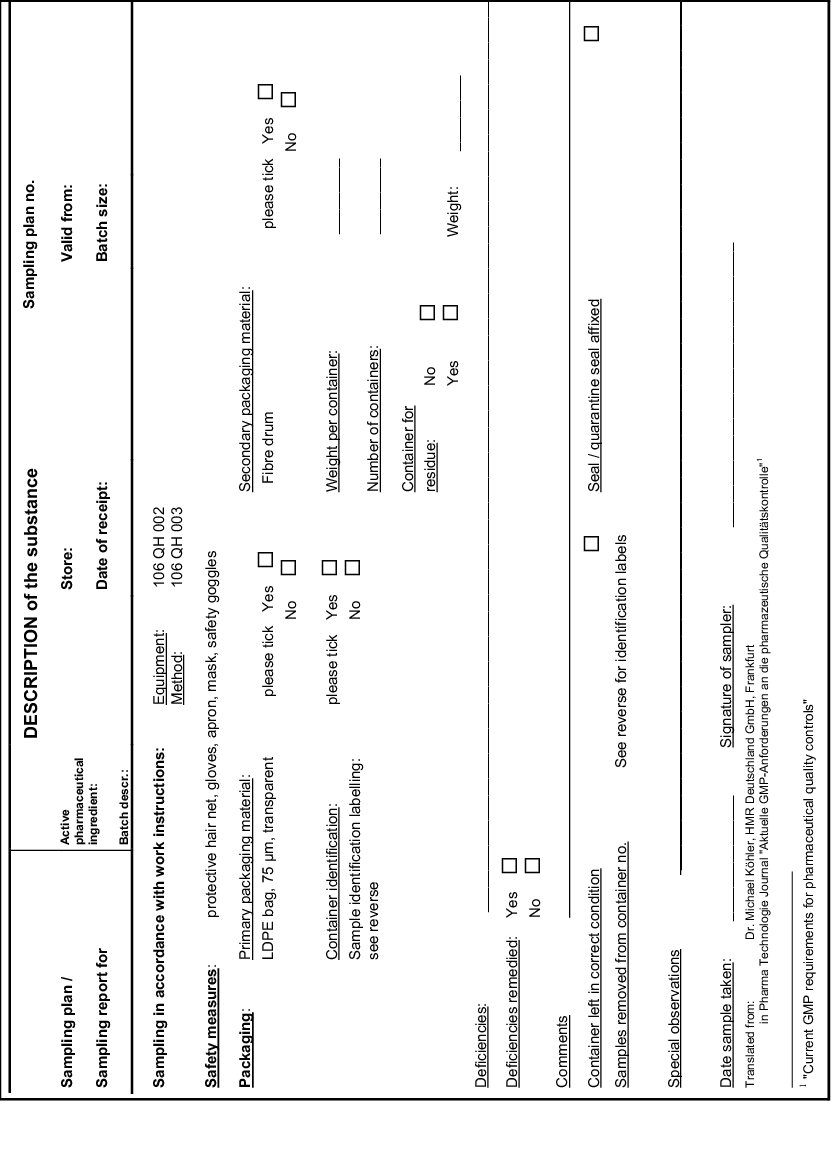
The method to be adopted for the sampling must be specified. This information shall include the number of samples (one or more) and the location where the sample was taken (e.g.at the bottom, below, outside, inside, on the top, in the center, at the wall). If several samples have been taken, it must also be specified whether or not the samples should be combined (combined samples or single samples). Combined samples are generally not permitted for container-based identity testing on a batch of starting material and for checking the content uniformity of a finished product. It is recommended to use forms for the sampling plan and the sampling report. An example (Köhler, 1997) of a combined sampling plan/sampling report has been reproduced in appendix 1 (see figure 16).
|
|
14.A.3 Notes for the sampling process
All the critical parameters must be taken into consideration when carrying out the sampling. If taking samples from a product in a tank truck, it is advisable to draw additional samples from the manhole and the discharge, to determine any particulate matters. If samples are taken from bags using dip probes, the possibility of contamination is particularly high (due to dust or germs). Ointment-based products must be melted before samples are taken.
14.A.3.1 Containers and identification labelling
| The sample containers must be clearly identified. This shall include: |
|---|
|
Once the sample has been taken, the container from which it was taken must be labelled accordingly (see figure 17) and sealed in such a way that the risk of a deterioration in the quality of the contents during storage is minimised (quality impaired). (See chapter 14.A.1.2 Equipment.)
14.A.3.2 Sampling report
The sampling shall be documented in the sampling report. The report shall cover all the items specified in the plan (instructions). This shall include clear description, quantity, batch number, date the sample was taken and (as an specific requirement for the USA) the name of the person who took the sample and the date that the sample for analysis was received in the quality control laboratory. A final requirement is to ensure that a sufficient amount of material is available so that all the tests can be carried out.
14.A.3.3 Reference samples
The number of reference samples required must be taken into consideration when taking samples. In general, the reference sample must be large enough to allow two complete analyses to be repeated.
Practice has shown that 2 types of reference sample can be used. A "sample A" is used for analysis within the specifications provided. A second "sample B" shall be specially protected and shall be touched under no circumstances. This sample shall be used as an internal reserve sample in the event of unforeseen circumstances (e.g. for settling legal disputes). A decision on this matter will be taken from an authorised department within the company.
Reference samples must be stored for at least 1 year beyond the expiration date for each batch. Finished products should be stored in their final packaging under the recommended storage conditions. For the USA, the following shall also apply: reference samples from representative batches (e.g. one batch per quarter) must be visually inspected at least once a year to ensure that they are being stored properly and that the sample material and container seal are in good condition. The inspections and their results must be recorded. If indications of not impaired quality should appear, investigations must be carried out to establish the cause.
Starting materials should be stored for 2 years after their last use if stability permits, excluding solvents, gases and water.
Summary The sampling process is relevant to all areas of pharmaceutical production and forms the basis of decisions relating to product quality. The personnel must be specially selected and regularly trained. The premises and the equipment must satisfy clearly defined requirements which exclude the possibility of contamination and promote ease of cleaning. Before sampling begins, sampling plans must be drawn up, according to which the sampling will then be carried out. The sampling must be neatly and comprehensively documented. Errors committed during sampling (e.g. through contamination) have far-reaching consequences. They cast doubt over the value of the ensuing stages in the quality control process; in addition, the subsequent trouble shooting procedure has proven to be very time-consuming and problematic. |
