Microbiological monitoring
Here you will find answers to the following questions:
|
It has become generally accepted that the manufacture of pharmaceutical preparations must take place under controlled microbiological conditions. Microbiological monitoring is therefore indispensable. The aim of the monitoring is to detect deviations from the validated state. Monitoring is designed to demonstrate that the process is under control.
12.G.1 Sources of contamination
Because the medicinal products come into contact either with air in the room or, under laminar flow units, with suitably purified air, air is often viewed as the main source of contamination. This is also reflected in the GMP guidelines which in places include extremely detailed requirements for the air system. For the manufacture of sterile products, the EU-GMP-Guideline (Annex 1: Manufacture of Sterile Medicinal Products) states that the air microbial count should be determined during operation in clean areas. The results should be taken into account when releasing a batch.
Surfaces are also relevant as possible sources of contamination. In the first instance, one must naturally view parts that come into contact with the product as particularly critical for product contamination, as organisms can be directly transferred here. However, there is no such thing as a non-critical point, as even here we must anticipate contamination via the air. For these reasons, all areas should be examined, not only those parts of the machine that come into contact with the product.
If the room fittings are defective (unsealed ceilings and windows, poorly equipped air lock and overpressure systems, etc.), massive invasions of organisms must be anticipated. Even the smallest scratches and cracks in the wall or floor covering give organisms a good chance of surviving disinfection measures and potentially developing into colonies. Particular attention must therefore be paid to the coating of floors and walls, as the paint and wall or floor covering will determine the success or otherwise of disinfection measures (see chapter 3.E Construction elements). The EU-GMP-Guideline gives very detailed specifications for structural fittings.
Because people are involved in nearly all production steps, particular attention must be paid to personnel hygiene. We must assume that personnel represent a main source of contamination. This is understandable when one considers the results of relevant investigations. A human being carries numerous organisms; not only aerobes, but also nearly as many anaerobes. Organisms are transferred to the product directly if, for example, an employee comes into contact with the medicinal product or indirectly, if the organism is first released into the air and then lands on the medicinal product.
Due to their nature, the usual measures for microbial prevention (sterilisation, disinfection) are not suitable for people, or only to a limited degree. One is generally limited to a hand disinfection here. Alongside this, suitable protective clothing in particular has an extremely important effect on organism release from people. Annex 1 of the EU-GMP-Guideline therefore contains very detailed specifications for protective clothing for cleanliness grades A to D. As well as wearing the clothes correctly, it is important to change them regularly as organisms penetrate through the material after a certain gestation period. It is particularly important to change gloves regularly to catch even the smallest amount of unnoticed damage (see chapter 11.B Personnel hygiene).
Due to the risk of the medicinal product being contaminated by the environment, monitoring must include the following areas:
- Air
- Surfaces
- Personnel
12.G.2 Room classification
If you wish to monitor the quality of a room, you need standards. Annex 1 of the EU-GMP-Guideline and the Microbiological Evaluation of Clean Rooms and Other Controlled Environment chapter of the USP recommend limits against which to evaluate the quality of the room. However, these are only guidelines. Alert and action levels (see chapter 12.G.3 Monitoring program) must be determined for the specific unit (figure 12.G-1).
| Area |
Required for |
|---|---|
Critical |
Sterile preparations - sterilised in sealed final containers
|
Sterile preparations - aseptic preparations
|
|
Critical |
Sterile preparations - aseptic preparations
|
Controlled |
Sterile preparations - sterilised in sealed final containers
|
Sterile preparations - aseptic preparations
|
|
Area with |
Sterile preparations - sterilised in sealed final containers
|
Sterile preparations - aseptic preparations
|
In order to determine useful levels, it is first necessary to divide the manufacturing areas into different room grades according to the type of product and production stage. Stricter room requirements are selected for preparations with high cleanliness requirements. We base our division of manufacturing areas on the room grades in Annex 1 of the EU-GMP-Guidelines (see chapter 3.D Room classes).
It is possible to compare the EU Guidelines (Annex 1: Manufacture of Sterile Medicinal Products), the FDA Aseptic Guidance and US Guidelines (USP) in terms of the particle limitation in the bodies of rules (see figure 12.G-2).
12.G.3 Monitoring program
Implementation of monitoring must be set down in writing. It is advisable to draw up the monitoring program in the form of a standard operating procedure. This standard operating procedure should include:
- Level
- Methods/equipment
- Frequencies
- Measures in the event of deviations
- Sampling (responsibilities)
- Processing (responsibilities)
- Sampling points
- Documentation
The monitoring SOP should also define where the documents (raw data, reports, deviation documentation, etc.) are stored. This is particularly important for any audits, as it is necessary to make documents available within a short space of time. Other details (e.g. incubation, culture media, etc.) should be described in the relevant laboratory SOPs.
12.G.3.1 Limits (level)
As mentioned above, alert and action levels should be determined for the specific unit. The EU-GMP-Guideline also requires that procedures for countermeasures are specified for the event that these limits are exceeded.
By alert level, we mean the limit above which there may be possible problems. Corrective measures are not necessarily required. If action levels are exceeded, one must assume that the process may be out of control. Investigations must be carried out to check this. Corrective measures may be required and their success should be checked (see chapter 12.G.6 Measure if levels are exceeded).
Inspectors expect levels to be determined in consideration of previous monitoring results and the room qualification. If you determine the level "by feeling", you lay yourself open to the accusation of making it too easy by setting the level too high. This is particularly the case for alert levels.
| FDA Aseptic Guidance |
USP |
EU-GMP-Guideline |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Operation room |
Operation room |
Class |
Quiet room |
Operation room |
|||||||
Clean Area |
ISO |
³ 0.5 mm |
Class |
Particles equal to and larger |
Max. permissible number of particles/m³ |
||||||
U.S. |
m3 |
ft3 |
0.5 mm |
5 mm |
0.5 mm |
5 mm |
|||||
100 |
5 |
3,520 |
M 3.5 |
100 |
3,530 |
100 |
A |
3,500 |
1(e) |
3,500 |
1(e) |
B |
3,500 |
1(e) |
|||||||||
1,000 |
6 |
35,200 |
No equivalent |
||||||||
10,000 |
7 |
352,000 |
M 5.5 |
10,000 |
353,000 |
10,000 |
B |
350,000 |
2,000 |
||
C |
350,000 |
2,000 |
- |
- |
|||||||
100,000 |
8 |
3,520,000 |
M 6.5 |
100,000 |
3,530,000 |
100,000 |
3,500,000 |
20,000 |
|||
D |
3,500,000 |
20,000 |
Not determined |
||||||||
It is relatively common to use the average of the results from a relatively long period of time +2 s (alert level) and +3 s (action level) as the basis for calculating the level (s = standard deviation). This procedure is even suggested by the German supervisory authorities. However, with this type of calculation, values are based on a normal distribution and outliers and level exceedances are overevaluated. Results from environmental monitoring usually follow a Poisson or negative exponential distribution. This is allowed for in the following formulae:
|
C = average value of all measurements |
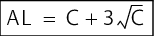
|
AL = alert level, C = average value of all measurements |

It is advisable to use the following procedure to implement these requirements for determining alert and action levels for daily monitoring:
- Action level If applicable, the level reported to the FDA must be taken into consideration (otherwise changes must be notified) and a regulation must also be established for handling the average values specified in Annex 1: Manufacture of Sterile Medicinal Products. To interpret the term "average value", we can use the recommendation from the FIP for air investigations (the average value should be smaller than 1 and be calculated from at least ten measurements) made in 1990.
- Alert level: It is advisable to calculate the level for the individual unit using the formulae stated above. However, infinitely high values are not acceptable.
Below are suggestions for various objects of investigation (air, surfaces, personnel) in the different cleanliness grades. The alert levels represent an upper limit.
Air
Annex 1 of the EU-GMP-Guideline recommends limits for microbiological contamination in order to assess the quality of the room for the manufacture of sterile products. Chapter <1116> and the FDA Aseptic Guidance also contain appropriate suggestions. In figure 12.G-5, there is an overview of the requirements and a suggestion for implementation. The action levels were based on the values from the bodies of rules.
| Grade |
FDA Aseptic Guideline |
USP |
EU-GMP- |
Proposal for implementation |
||
|---|---|---|---|---|---|---|
| Determination |
2004 |
1987 |
Recommended |
Alert level |
Action |
|
100 (ISO 5) |
||||||
Quantitative |
1* CFU/m3 |
<3 CFU/ |
<3 CFU/ m3 |
<1 CFU/ m3 |
- |
0 (1) CFU/ |
Settle plates |
1* CFU/4 h |
- |
- |
<1 CFU/4h |
- |
- |
1,000 (ISO 6) |
||||||
Quantitative |
7 CFU/m3 |
- |
- |
- |
- |
- |
Settle plates |
3 CFU/4 h |
- |
- |
- |
- |
- |
10,000 (ISO 7) |
||||||
Quantitative |
10 CFU/m3 |
- |
<20 CFU/ m3 |
10 CFU/ m3 |
3 CFU/m3 |
7 CFU/m3 |
Settle plates |
5 CFU/4 h |
- |
- |
5 CFU/4 h |
- |
- |
100,000 (ISO 8) |
||||||
Quantitative |
100 CFU/m3 |
<80 CFU/m3 |
<100 CFU/m3 |
<100 CFU/m3 |
50 CFU/m3 |
100 CFU/m3 |
Settle plates |
50 CFU/4 h |
- |
- |
50 CFU/4 h |
- |
- |
D (particle count not determined) |
||||||
Quantitative |
- |
- |
- |
<200 CFU/m3 |
200 CFU/m3 |
400 CFU/m3 |
Settle plates |
- |
- |
- |
100 CFU/4 h |
- |
- |
* Air monitoring samples of critical areas should normally yield no microbiological contaminants, ** Averages |
||||||
Surfaces (rooms/technical equipment)
In Annex 1 of the EU-GMP-Guideline, there are general limits for each cleanliness grade A to D, but no information about whether these are for personnel or surfaces and no differentiation according to location (table, wall, floor, etc.). As there is additional data available, these are certainly meant to be requirements for surfaces. We can assume that these are essentially surfaces close to the product and not floors (microbiological checks are made here to verify the disinfection procedures and not as part of the production monitoring). The USP and FDA Aseptic Guidance (for product contact only) also contain relevant suggestions. In figure 12.G-6, there is an overview of the requirements and a suggestion for implementation.
| Grade |
FDA Aseptic Guideline |
USP |
EU-GMP- |
Proposal for implementation |
||
|---|---|---|---|---|---|---|
| Determination |
2004 |
1987 |
Recommended |
Alert level |
Action level |
|
100 (ISO 5) |
||||||
Product contact |
0*CFU/ |
- |
- |
< 1 CFU/ |
- |
0 (1) CFU/ |
Surfaces |
- |
- |
3 CFU/25 cm2 |
- |
2 CFU/ |
3 CFU/ |
Floor |
- |
- |
3 CFU/25 cm2 |
- |
3 CFU/25 cm2 |
5 CFU/ |
10,000 (ISO 7) |
||||||
Surfaces |
- |
- |
5 CFU/ |
5 CFU/ |
2 CFU/ |
5 CFU/ |
Floor |
- |
- |
10 CFU/ |
- |
5 CFU/ |
10 CFU/ |
100,000 (ISO 8) |
||||||
Surfaces |
- |
- |
- |
25 CFU/ |
25 CFU/ |
50 CFU/ |
Floor |
- |
- |
- |
- |
||
D (particle count not determined) |
||||||
Surfaces |
- |
- |
- |
50 CFU/ |
100 CFU/ |
200 CFU/ |
Floor |
- |
- |
- |
- |
||
* Critical surfaces that come into contact with the sterile product should remain sterile throughout an operation, ** Averages |
||||||
Personnel
As already mentioned, Annex 1 to the EU-GMP-Guideline contains recommended limits for monitoring personnel in cleanliness grades A and B. Chapter <1116> also contains relevant suggestions. The FDA Aseptic Guidance points out that production personnel should wear non-contaminated gloves and clothing in the sterile room during intervention. In figure 12.G-7, there is an overview of the requirements and a suggestion for implementation.
| Grade |
FDA Aseptic Guideline |
USP |
EU-GMP- |
Proposal for implementation |
||
|---|---|---|---|---|---|---|
| Determination |
2004 |
1987 |
Recommended |
Alert level |
Action level |
|
100 (ISO 5) |
||||||
Gloves |
0*CFU/25 cm2 |
- |
3 CFU/25 cm2 |
< 1 CFU/ |
- |
0 (1) CFU/ |
Clothing |
0*CFU/25 cm2 |
- |
5 CFU/25 cm2 |
- |
||
Hood |
0*CFU/25 cm2 |
- |
5 CFU/25 cm2 |
- |
||
10,000 (ISO 7) |
||||||
Gloves |
- |
- |
10 CFU/25 cm2 |
5 CFU/ |
2 CFU/ |
3 CFU/ |
Clothing |
- |
- |
10 CFU/25 cm2 |
- |
5 CFU/ |
10 CFU/ |
Hood |
- |
- |
20 CFU/25 cm2 |
- |
5 CFU/ |
10 CFU/ |
* An ongoing goal for manufacturing personnel in the aseptic processing room is to maintain contamination-free gloves and gowns throughout operation, ** Averages |
||||||
12.G.3.2 Methods and equipment
This item in the monitoring program must clearly determine the method with which the investigation is to be carried out. Details for sampling should be regulated in laboratory SOPs.
Microbiological testing of air
Airborne organisms do not generally levitate but are bound to particles. However, all attempts to establish a reproducible relation between organisms and particles have failed to date. This means one must carry out microbiological investigations in order to make any conclusions about the air microbial count. There are both qualitative and quantitative methods available for checking the air microbial count (see figure 12.G-8).
Qualitative procedure
- Settle plates
Quantitative procedures
- Filtration
- Impaction
- Impingement
For the settle plate procedure, petri dishes containing a suitably solid medium are left open in a room for a certain time (30 minutes to 2.5 hours; EU-GMP-Guideline Annex 1: 4 hours). The organisms that settle on the agar surface in this time can be counted following incubation. Even though the EU-GMP-Guideline contains levels for this method, quantitative conclusions cannot be drawn, as they depend on numerous coincidences. This method should therefore only be used in conjunction with quantitative methods. This is also referred to in the FDA Aseptic Guidance.
|

The FDA Aseptic Guidance also states that, as part of the method validation with settle plates, no drying out must be allowed to occur, as this can influence the recovery of organisms. For these types of conformance tests, it is certainly not sufficient to use the USP reference strains, otherwise isolates from the unit being checked would also be required. This requirement to prevent settle plates drying out should be checked for the selected settle plates. There are obviously differences between the cultural medium plates of different manufacturers. A fill quantity of 30 ml/plate is generally more favourable than 20 ml. Such tests can be carried out by weighing. This indicates that, to all intents and purposes, plates that are available on the market show an extraordinarily low degree of drying (<2 %), even with the exposure time of four hours suggested by the FDA when stating the levels. Whether this would be accepted by the FDA as a prevention of drying out is questionable.
With the filtration method, the air is sucked through membrane filters (generally gelatine filters). Microorganisms are caught by these and can then be cultured. A main advantage of the filtration method is the possibility of taking isokinetic measurements (suction rate > flow rate) that are needed to be able to measure under LF.
The impaction procedure (air striking solid culture media) includes the Anderson sampler, popular in the USA, the Reuter centrifugal sampler (RCS) and the slit sampler (STA method). With these methods, the air is sucked in, accelerated and blown onto solid culture media. Due to their mass, the particles and any organisms bound to them remain on the culture medium. This technique is only able to determine a certain range of particle sizes. Whereas the Anderson sampler covers a very broad range of particle sizes with its cascade arrangement of six sieve plates, the RCS unit and the slit sampler aim to cover the range that is typical for the air (this varies, however, depending on the cleanliness grade of the room).
In the USA, the STA method is favoured by the FDA. In the Microbiological Evaluation of Clean Rooms and other Controlled Environment chapter of the USP, air levels are therefore also set with reference to the STA (Using a Slit-to-Agar Sampler or Equivalent). The use of equivalent samplers is expressly permitted.
With the slit sampler, the air to be tested is blown onto a rotating agar plate. This makes it possible to detect contamination in relation to time. According to the manufacturer, all particles greater than 0.5 mm are detected. Bacteria are sometimes smaller than this, but because they cling to particles they are reliably detected with this particle separation. The manufacturer states an exposure time of up to 90 minutes. This is in line with the requirements of FDA investigators to cover as long a period of time as possible. A test volume of 55 l/min is possible, which corresponds to 3.3 m3/h (maximum total testable volume: approx. 5 m3). However, special plates with a diameter of 15 cm are required. These are then analysed with a graded template for recognising temporal context. In order to save material, it has been suggested on various occasions that a plate can be used several times. This is of course possible, but the possibility of detecting temporal context is then lost.
The RCS method is very practical. The battery-operated unit works according to the impaction principle and allows a qualitative separation of airborne microorganisms from a sample volume of between 10 and 1000 litres. The flow of air enters the rotor from the front, is rotated by the fan blade and organisms in the air are separated onto the airborne organism indicator by centrifugal force. The air outlet is directed towards the rear, parallel to the unit, to prevent turbulence in the intake area. The sample volume at a speed of 6,100 rpm is approximately 50 l/min, whereby lighter or smaller particles and those that are somewhat further from the agar surface have enough time to settle in the gap between the inner part of the rotor and the agar surface. Because a sufficiently high suction rate is achieved and a measurement of 1 m3 is possible, the unit can be used under LF.
Finally, we should look at the impingement procedure (the collection of organisms in a liquid). With this method, a defined volume of air is sucked through a liquid or directly through a culture broth. Following sufficient mixing, the microorganisms present in the air are retained in the liquid, which can then be microbiologically tested for microbial content. Although the impingement procedure is often viewed as the "standard procedure" in scientific investigations, it is seldom used for routine testing as it is extremely expensive.
The question is, which method should one routinely use? Numerous comparative studies have shown that the results are extraordinarily dependent on the method. For this reason, the method used should always be stated for results and requirements. It is important that routine air testing is always carried out with the same method. This is the only way to detect changes in the air quality.
12.G.3.3 Microbiological testing of surfaces and personnel
If possible, a sample should only be taken from surfaces that come into contact with the product after production has finished, so as not to endanger the product. This is also the case for personnel, so that gloves are not contaminated.
The following methods are usually used today to test the microbial count on surfaces and personnel (see figure 12.G-9):
- Contact plate method with solid culture media
- Swabs
The following method is also used for personnel monitoring:
- Hand washing method (rinsing procedure)
|

Contact plate method with solid culture media
The contact plates have a convex agar surface (25 cm2 total area) that allows them to be pressed directly onto the surface to be tested. Agar films make it possible to test even very curved surfaces (e.g. in vessels). Agar films also have a convex agar surface (approx. 25 cm2 total area), so that they can also be pressed directly onto the surface to be tested.
When using contact plates of agar films, you must make sure that there is no condensate (the colonies could run together). They must also be checked macroscopically for any contamination. Only plates with an expiry date that is after the anticipated incubation period may be used. Once the sample has been taken, it is extremely important to clean the agar residue from the contact surface with sterile-filtered 70 % alcohol. This is the only way to ensure that agar residue does not improve microbial growth of possible contaminants on the tested surface.
When personnel are tested, gloves must not be disinfected immediately before samples are taken. The result would be massively distorted and is not therefore usable. This is also referred to in the FDA Aseptic Guidance.
Because the contact pressure and the contact duration should have a significant influence on the microbial yield, bioMérieux (bioMérieux Deutschland GmbH, Postfach 1204, D-72602 Nürtingen, Germany) have brought an aid for contact plates onto the market which standardises these two parameters (uniform pressure of 500 ± 50 g over a period of 10 ± 1 seconds).
Swabs
A swab consists of a rod to which a cotton wool pad or alginate pad is affixed. The surface can be wiped with this pad.
A damp swab (swab dampened with sterile physiological NaCl solution) is used to test dry surfaces. The sample material collected is then wiped onto casein soya bean digest agar. The top of the pad can then be cast into casein soya bean digest agar and both plates incubated.
A dry swab is used to test damp places (e.g. remains of cleaning water on the floor). The sample material collect is then wiped onto casein soya bean digest agar. The top of the pad is then put into casein soya bean digest broth for concentration for evidence of pseudomonas aeruginosa. The plate and test tube are then incubated.
The alginate swab gives the best microbial yield. It should be used especially when only small microbial counts are expected. The pad on the alginate swab is first soaked in a sodium-citrate buffer. The solution or suspension is used to carry out a blend test in casein soya bean digest agar. The remaining pad is cast into casein soya bean digest agar and both plates are incubated.
Hand washing method (rinsing procedure)
The FIP (Fédération Internationale Pharmaceutique) describes this interesting method. Both hands are washed in 1 litre of sterile saline solution, with the addition of a disinfectant neutralising substance if necessary. The microbial count is measured by filtering portions of the wash fluid.
It is certainly expedient to add 0.1 % of Tween 80 to the wash fluid to ensure better cleaning. The microbial count on the hands can be determined by filtering the wash fluid and incubating the filter. The FIP suggests 30 CFUs as the level for both hands. However, this is high for a gloved hand and not acceptable to an inspector. If you wish to use this method, you should carry out a comparative study of hand contact with an acceptable result and the hand washing method level.
In literature, this hand washing method appears to be similar to the rinsing procedure of horizontal surfaces with subsequent filtration. In my opinion, this procedure is not particularly suitable, as it is virtually impossible not to create puddles, which entail their own problems with water organisms (in particular pseudomona aeruginosa).
Culture media
According to suggestions in the USP, it is advisable to use casein soya bean digest agar. As this medium is prescribed for the total microbial count determination of final products, it is assuredly well suited for this purpose. Inactivating agents should be added to the medium so that any growth-neutralising substances picked up are inactivated. This is naturally always the case for surface testing, as disinfectant residues must be expected on the tested surfaces, as well as for contact by a gloved hand, for example. An inactivating additive can also be useful for air testing, for example, if antibiotic dust is to be expected.
If you purchase ready-to-use contact plates, you will get at least polysorbate and lecithin as neutralisers (e.g. contact plates from BBL), plus perhaps L-Histidine (e.g. Count-Tact plates from bioMerieux, GK-HYCON organism indicator from Biotest and contact plates from Heipha). If you are working with swabs, you should be aware that disinfectant residue can be picked up during the swabbing procedure, which can make it necessary to add suitable substances to the medium. In figure 12.G-10, there is a selection of neutraliser combinations recommended by the German Society for Hygiene and Microbiology (Deutschen Gesellschaft für Hygiene und Mikrobiologie - DGHM).
| Recommended inactivation mixtures |
|---|
|
Particular attention must be paid to avoiding bringing organisms into the sterile room on the exterior of the test material. It is often attempted to disinfect the exterior by spraying with a disinfectant. This should be done with care so as not to influence the growth promotion of the medium with disinfectant residue. This procedure must be validated. It is preferable to use double-wrapped sterilised plates or strips.
As with all microbiological testing, the culture medium should be checked for growth promotion. For this, the agar strips or plates must be inoculated with up to 100 CFU of the usual test strain of USP or Ph. Eur. 5 (see figure 12.G-11 and chapter 12.H Test for sterility).
| Organism group |
Organism |
Strain, e.g. |
|---|---|---|
Aerobes |
B. subtilis |
ATCC 6633 |
Anaerobes |
Clostridium sporogenes |
ATCC 11437 |
Fungi |
Candida albicans |
ATCC 10231 |
Incubation
The FDA Aseptic Guidance states the following as incubation conditions:
- Aerobes: 48-72 hours at 30-35 °C
- Yeast and mildew: 5-7 days at 20-25 °C.
If the FDA suggestion is followed, it means that two samples must be taken (one sample for 48-72 hours at 30-35 °C and one for 5-7 days at 20-25 °C). By way of compromise, a single sample can be taken and first incubated for 5(-7) days at 20-25 °C and then (after an interim reading) 48(-72) hours at 30-35 °C. Both suggestions entail a relatively high amount of effort for carrying out the test and even more documentation (two readings). If previous incubation has only ever been at one temperature, incubation at two temperatures means an interruption in the trend recording.
If you decide to incubate at only one temperature (in accordance with the FIP suggestion, for example: 5 days at 30 ± 2 °C), the equivalence of the procedure must be proved in extensive tests. This excludes all the problems mentioned above, but appropriate validation studies must be carried out to also include isolates. This type of procedure is acceptable, but the FDA expressly states that, for environmental monitoring, microbiological test methods other than the traditional ones (e.g. USP) may be used if it has been proven that these methods provide equal or better results. If only one incubation temperature has been used previously, this procedure is to be recommended, in spite of the validation requirements, as the trend recording can be continued.
The question of the need for anaerobic incubation of the monitoring samples comes up again and again. If we consider that anaerobic organisms occur in humans in the same quantities as aerobic organisms, this need seems to be understandable In the context of media fills (see chapter 12.E.5 Culture medium filling (Media Fill) and chapter 12.H.9 Culture media controls), FDA investigators ask for information about anaerobes. If this is not available, they request media fills with thioglycolate medium. By comparing the level exceedances in the tests of samples with aerobic and anaerobic incubation, we were able to determine that anaerobic incubation only rarely provided additional information.
During media fills, we therefore carry out anaerobic incubation on a second sample in some places in order to show that we do not have any problems with anaerobes and that culture medium filling using thioglycolate medium is not necessary.
If solid culture media (agar plates, contact plates, agar strips, etc.) are to be incubated in anaerobic conditions, this is carried out in large quantities in an anaerobic jar. If only a small number is to be incubated, this can also be done with anaerobic bags (e.g. GENbag gas generators by bioMérieux, Nürtingen). It is important that the anaerobic ratios can be actually achieved on the agar strips, in the plate, etc. Agar strips must not be adhered under any circumstances. If cams are present, hooks must be placed in the plates to enable gas exchange.
Method validation
Methods for microbiological monitoring must be validated. This is referred to in the FDA Aseptic Guidance. Inspectors, in particular those from the FDA, will also want to see the relevant documentation. Unfortunately, it is not always so easy to carry out suitable tests, as often the necessary technical equipment (such as an aerosol chamber for producing a microbe cloud) is not available. However, it is possible to carry out the expected validation up to a certain degree and to submit this if necessary.
|

In surface testing methods, it is primarily the recovery rate that is questioned. This means that the surfaces must be artificially contaminated and then tested to see how many organisms can be detected with the selected method. Here, we will generally attempt to answer this question with data from literature. In figure 12.G-12, there is a summary of relevant tests carried out by the author. The source quoted also contains the associated statistical data.
For air testing methods, the FDA Aseptic Guidance requires that active collectors are calibrated and, when using settle plates, that the exposure conditions are optimised to the unit with relevant testing. In practice, the relevant data on the air volume tested and on the air velocity (important when using the equipment under LF) should be requested from the equipment manufacturer. In the laboratory, this data should be checked regularly (six-monthly) if possible. If this is not possible, the equipment must be sent in and must have the relevant test certificates. Every batch of agar strips, contact plates, etc., must be tested for growth promotion (see Culture media page 14). If culture media are not covered at the end of the incubation, it must be demonstrated that organism growth would not have been possible by dripping test organisms onto them (maximum 100 CFUs, e.g. of staphylococcus aureus ATCC 6538).
It is also important to validate the air lock process. For this, inoculation with organisms (complete range of organisms, see Culture media page 14) after the air lock should show that nothing is growing on the medium in the H2O2, formalin, etc. in the material lock and thus that the growth promoting properties are still present to a sufficient degree.
12.G.4 Sampling
It is not sufficient to describe sampling procedures, etc. here (see chapter 14.A Sampling). It is particularly important to determine the responsibilities for sampling and, if necessary, also for transportation from the unit to the processing laboratory. This is the only way to ensure that the monitoring runs smoothly.
It is similar for the processing of the samples, in that the responsibilities must be clearly defined. It must be ensured that no samples are left unprocessed (see chapter 14.A.2 Sampling plan (instructions)).
12.G.4.1 Frequencies
For a monitoring program to be compiled, the testing frequencies must be considered. There are guidelines for this in both the European and the American directives. Whereas in Europe, testing at the end of a process is sufficient, the FDA Aseptic Guidance requires that monitoring takes place during the manufacturing and filling activities. If the operation has several shifts, the daily monitoring should also cover every shift. This is interpreted generally to mean testing during manufacturing, spread out over the process. FDA investigators also require concentration on the actual filling area.
Air
The supplement to the EU-GMP-Guideline for the manufacture of sterile medicinal products contains specific information about testing frequencies. Microbiological monitoring is required during production for aseptic processes, in the same way as for FDA production. FDA investigators expect general measurements spread out over the production process. By contrast, the supplement to the EU-GMP-Guideline for the manufacture of sterile medicinal products is satisfied with monitoring based on the processing step: air during or at the end of production. From now on, FDA investigators require concentration on the actual filling area, whereby they would like a long period of time covered for air. In figure 12.G-13, there are frequencies for sterile areas that have been accepted by the FDA.
| Frequencies for air testing in sterile areas |
|
|---|---|
| Sampling points |
Frequency |
Critical area A |
EU production: for products to be manufactured aseptically, per batch after the critical processing step. A single measurement if several batches are manufactured per day. |
US production: at the start of a batch, then every 2 hours (min. 2) |
|
Critical area B |
EU production: after the critical processing step and during use if there is no increased risk caused by the measurement. |
US production: at the start, in the middle and at the end of production of a batch |
|
Controlled area C |
operation room - daily to quarterly (depending on the production frequency or room usage) |
Area with |
Operation room - monthly to quarterly |
Surfaces
The supplement to the EU-GMP-Guideline for the manufacture of sterile medicinal products contains information about testing frequencies for surfaces too. Microbiological monitoring is required during production for aseptic processes, in the same way as for FDA production. FDA investigators expect general measurements spread out over the production process. By contrast, the supplement to the EU-GMP-Guideline for the manufacture of sterile medicinal products requires that the sample is taken at the end of a critical processing step so that the drug product is not endangered. In figure 12.G-14, there are frequencies for sterile areas that have been accepted by the FDA.
| Frequencies for surface testing in sterile areas |
|
|---|---|
| Sampling points |
Frequency |
Critical area A |
|
Surfaces |
|
|
|
Wall |
|
|
|
Floor |
|
|
|
Critical area B |
|
Surfaces |
|
|
|
Wall |
|
|
|
Floor |
|
|
|
Controlled area C |
|
Surfaces |
|
Wall |
|
Floor |
|
Area with requirements D |
|
Surfaces |
|
Wall |
|
Floor |
|
Personnel
The supplement to the EU-GMP-Guideline for the manufacture of sterile medicinal products also contains information about testing frequencies for microbiological checks of personnel. Microbiological monitoring is required during production for this, in the same way as for FDA production. The sample should again be taken at the end of the process in order not to endanger the drug products. In figure 12.G-15, there are frequencies for sterile areas that have been accepted by the FDA.
| Frequencies for personnel testing |
|
|---|---|
| Sampling location |
Frequency |
Critical area A |
|
EU production: |
|
Hand |
|
Forearm |
|
Hood |
|
USA production: |
|
Critical area B |
|
EU production: |
|
Hand |
|
Forearm |
|
Hood |
|
USA production: |
|
12.G.5 Sampling points
The critical points must be considered in the first instance when determining the sampling points. The results of the initial monitoring when setting up a sterile room must also be taking into consideration when determining the sampling points. For example, in the filling station, testing is certainly expected near to the filling needle. If other problem areas come to light later (for example, during investigations), these must also be included in the routine monitoring.
However, you should beware of testing as many points as possible. This would only produce a vast quantity of data (and cost) without any additional gain of information. In the very compact facilities that are usual nowadays, one or two sampling points in the filling machine is sufficient - and likewise in the surrounding area. The PDA Technical Report 13 Revised Fundamentals of an Environmental Monitoring Program, which was mentioned earlier, contains a table of examples of sampling points (see figure 12.G-16).
| Examples of sampling points (PDA 2001) |
|
|---|---|
| System |
Point |
Air (filling line) |
|
Room air |
|
Surface (room) |
|
Surface (equipment) |
|
Personnel on the filling line |
|
Laminar air flow |
|
The sampling points must be clearly determined (e.g. not only "on the table", but "30 cm at the centre of the table"). By way of description, it is also useful to have a plan of the sterile room with sampling points plotted. This facilitates discussions in the event of an inspection.
12.G.6 Measure if levels are exceeded
The measures for this should only be referred to in general terms in the monitoring SOP. Details should be set down in a separate SOP. It is not simply a case of surveying and logging the results. If necessary, corrective measures must be implemented and appropriately documented. First, an investigation plan (testing plan) must be drawn up and then, following successful testing, an investigation report must be compiled.
Investigation plan
The following must be checked as part of this investigation:
- Results
- Extent of the problem
- Influence on the products
- Are quarantine measures required
- Follow-up testing
And the following must be implemented:
- Investigations to find the cause and
- Investigation of measures to show their success
It is important to first ask whether this is a question of a deviation in production hygiene or possibly an incorrect laboratory result.
If there is no error in analysis of the microbiology, the question of the influence on the product quality is of vital importance. The product should be quarantined if the deviation is relevant to the product. There is generally no relevance to the product if there are deviations in the personnel lock, for example. If the deviation is relevant to the product, investigations must be carried out to determine whether the product quality is actually affected.
In PDA Technical Report 13 Revised Fundamentals of an Environmental Monitoring Program (2001), there is a useful summary of corrective measures in the event of deviations (see figure 12.G-17).
| Typical corrective measures if levels are exceeded (PDA 2001) |
|
|---|---|
| System |
Corrective measures |
Room air/ |
|
Facility surfaces |
|
Personnel |
|
If necessary, additional testing must be carried out to find the source of the organisms. If the action level is exceeded, the organisms must always be identified. This will often provide information about the source of the organisms. Once the measures have been carried out, appropriate testing must demonstrate that they were successful and that the area or process is under control again.
Investigation report
Once the investigations are complete, an investigation report must be drawn up, documenting the following:
- Investigations
- Measures
- Review by management
- Distribution to affected personnel
- Any future corrective measures
12.G.7 Organism identification
Organisms discovered must be identified as part of the monitoring, at least if the levels are exceeded. The hope is that this will provide information about the source. Unfortunately, conventional methods for identifying organisms are not always enough to detect clear contamination contexts. This is expressly stated in the new, harmonised sterile testing chapter of the Ph. Eur. and the FDA Aseptic Guidance also contains reference to this. Because most microbiological laboratories do not have the facility for molecular isolate typing, one generally has to be satisfied with conventional methods. But even with these results, it is possible to draw certain conclusions about the possible source of the organisms based on the type. Relevant specialist literature is also helpful, such as Mitscherlich, E. and E.H. Marth, Microbial Survival in the Environment, Springer-Verlag, Berlin, Heidelberg, New York, Tokyo (1984). There is also help on the Internet at the address http://141.150.157.117:8080/prokPUB/index.htm.
In environmental monitoring, one must anticipate very varied proportions of the different organism groups:
- Gram-positive rods (floor, dust): 10-20 %
- Gram-positive cocci (human and animal origin): 80-85 %
- Gram-negative rods (water, floor): 5-10 %
- Yeast and mildew: 2-5 %
The following section presents some possibilities for the examination procedure to identify isolates (bacteria) in environmental monitoring. It is extremely important to note that this type of work is covered by the infection protection legislation and must therefore be notified to the responsible authorities (epidemic unit of the responsible regional government) and approved by them.
Normally, not all colonies that have exceeded a level are identified and one must be satisfied with the identification of all morphologically different colonies. It is extremely important to run one, or often even two, passages on blood agar to get a single colony. The organisms are revitalised by this and acquire the potential of carrying out their typical biochemical reactions. They should be cultured in a fractionated smear so that pure cultures can be obtained for the subsequent work. The individual colonies are checked first (colour, size, shape, hemolysis, any odour, metallic gloss, etc.) and the results are logged. The subsequent procedure is determined with a gram preparation (figure 12.G-18 ).
|

The following diagrams show examination procedures for gram-negative rods (figure 12.G-19), gram-positive rods (figure 12.G-20), gram-negative cocci (figure 12.G-21) and gram-positive cocci (figure 12.G-22). The bioMérieux api identification system is stated as an example. The other systems available on the market can of course also be used here.
|

|

|

|

Of course, it is not sufficient to only consider bacteria. If problems with fungi occur, a distinction must first be made between yeast and mildew. If a more precise identification is required, one must generally turn to specialists as, for mildew at least, there are no reliable identification systems on the market.
As every organism found cannot generally be identified for reasons of capacity, one must decide when it is desirable to identify the organisms and to what extent. According to the FDA Aseptic Guidance, organisms should be identified to species level as far as possible and - if appropriate - to genus level.
If the action level is exceeded in cleanliness grade A and B, organisms should certainly be identified to species level if possible. If the alert level is exceeded then identification to genus level is usually sufficient. If there is a suspicion that special organisms are present, e.g. spore formers or pseudomonads, these should also be identified.
In cleanliness grade C, identification to genus level is generally sufficient if the action level is exceeded. If there is a suspicion that special organisms are present, e.g. spore formers or pseudomonads, these should also be identified here.
For isolates from grade D, colonies that occur particularly often should be identified. Gram staining is usually sufficient.
Summary When manufacturing and filling sterile drug products, all possible sources of microbial contamination from the environment must be controlled. This primarily concerns air, surfaces and personnel. A monitoring program must be established for this, in which levels, sampling points, frequencies, methods/equipment and measures in the event of deviations, amongst other things, must be regulated. These individual points are explored and suggestions are made for practical implementation. |
