Reworking
Here you will find answers to the following questions:
|
The reworking of products or semi-finished products (bulk) is possible for a number of reasons. In principle, a differentiation may be made between the reworking of
- products that have been rejected and
- products that have not been rejected
11.L.1 Reworking rejected products
The EU GMP Guideline expressly permits the reworking "[...] of rejected products [...]". (5.62 EU GMP Guideline) Conditions under which this kind of reworking may be carried out are cited (see figure 11.L-1).
| Reworking rejected products |
|---|
|
An important question to ask when assessing reworking is whether the current deviation occurs regularly. Deviations should rarely occur. They can occur in isolation, e.g. due to mechanical defects. Assuming that the manufacturing process has been validated, this kind of reworking is not generally required. Where deviations occur more frequently, it can be assumed that the process or process chain is not stable.
The procedure must be defined in the form of a process instruction that states which areas or persons are responsible for the various steps. figure 11.L-2 shows an example.
|

The result of the research is presented in the deviation report in which the order of events and causes are documented (see chapter 11.K Deviations). In this example, the results are compiled by the company's galenic unit. Knowledge about the causes of the deviation allows a proposal for a reworking procedure to be compiled which in this case is also put forward by the company's galenic unit. The evaluation of the proposal based on the risk assessment is carried out by the head of quality control with the support of the relevant specialist departments. If the result of the assessment is positive, manufacturing instructions may be compiled. These must be authorised by persons in the relevant responsible positions. The reworking itself is then subsequently carried out.
As far as the background in relation to the quality of the finished product is concerned, the evaluation of the risk associated with reworking must sometimes be regarded as critical. Economic requirements must be considered in relation to quality requirements. The decision is to be documented - for example, in the deviation report together with the reworking proposal. The batch record of the starting batch as well as the reworked batch must carry a reference to this report or even include the report to guarantee traceability of the data.
|

"The quality control department should assess whether a finished product [...] must be subjected to additional analyses" (5.64 EU GMP Guideline). This is a consistent approach as the qualitative assessment of a product should be considered separately in such instances. It must be demonstrated that the reworking procedure does not adversely affect the quality of the product, e.g. as with the effects of heat when crushing solids. The additional tracing of possible degradation products may subsequently be necessary.
Example
The sequence is illustrated using the following example for the production of solid dosage forms.
|
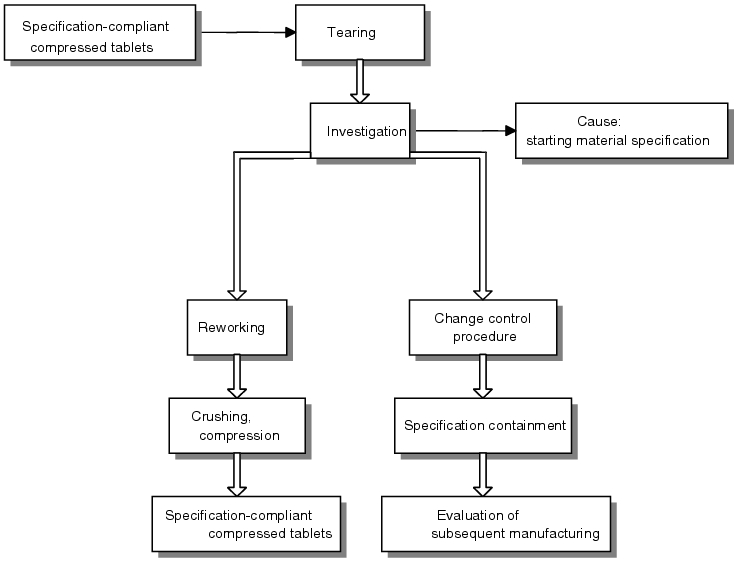
- Description of situation
Using the set tablet height as the starting point, the breaking strength was kept at the upper specification limit during compression. In this case, the tablets were overpressed as a result of the pressing force selected, i.e. no additional increase in breaking strength was achieved through the application of excess pressing force. Formation of cracks in the tablets became apparent as a relaxation effect following a standing time of approximately 1 hour. The compressed tablets manufactured could not be processed further due to the stability risk. All values documented (tablet height, breaking strength, pressing forces) were in conformance with the specifications. - Report
It was possible to establish a connection through the research initiated between raw material specifications (in this case grain sizes) and the required pressing force. Although the specification for the maximum grain size was satisfied (maximum particle size), it emerged that the grain size distribution (not part of the specification) was different to the development data (considerably higher fine-grained fraction, smaller average grain size). - Perspective
It was therefore possible to narrow the specification for subsequent batches to achieve regulatory compliance for manufacturing process values. The tablets were reworked by crushing them and then pressing them once again.
11.L.2 Reworking of products that have not been rejected
„The recovery of all or part of earlier batches which conform to the required quality by incorporation into a batch of the same product [...] should be authorised beforehand. This recovery should be carried out in accordance with a defined procedure [...]. The recovery should be recorded." (5.63 EU GMP Guideline).
In this case, batches or part batches are reworked and then used to manufacture further batches. In so doing, the reworked material should be handled in the same way as a starting material, i.e. determination of release status following quality control, traceability through identification (e.g. individual material number, batch designation) must be considered. For recycling, authorised manufacturing instructions must exist in which the precise allocations of the reworked quantities are documented. The reworking and reintroduction of these quantities into manufacturing processes must be validated.
The reworking of products that have not been rejected may include, for example, the removal of tablets from the blisters and then their repackaging. In this case, the blisters are initially removed from the folding cartons so that the tablets can then be mechanically pressed out of the blisters. Before the bulk obtained is packaged, this must undergo a visual inspection prior to approval. This involves testing attributive features such as chipped or broken edges etc. in a larger random sample (see chapter 13.A.5 Packaging material testing) as the tablets may be damaged when they are removed from the blisters. If the set limits are exceeded, sorting processes will be required. The tablets are to be repackaged following approval, in doing so, the usual precautions must be taken to prevent confusion and cross-mixes. Due to the high risk of misidentification, it must be ensured that all the packaging materials of the original batch are gathered up and destroyed to exclude the possibility of a cross-mix (see chapter 13.B Packaging process). During repackaging, care should be taken to ensure that the expiration date has not changed. The running time of the product cannot be extended by repackaging.
Summary Reworking may be carried out for products that have been rejected as well as those that have not been rejected following approval. The reworked batches are subject to stringent quality controls. It must be demonstrated that reworking has no negative influence on the product concerned. |
