|
Here you will find answers to the following questions:
|
1 Regulative Aspects
Process validation is a fundamental component of the quality assurance system used by pharmaceutical manufacturers. It should verify that the procedures and processes used in drug product manufacturing are suitable for their purposes and guarantee that the drug product manufactured is of the required quality. A procedure is an established way of carrying out an activity. A process is a set of methods and actions that interact to convert inputs to outputs. Process validation is a basic factor for drug product safety.
1.1 Legal requirements for drug products
The holder of a manufacturing authorisation must ensure that manufacturing and analysis are carried out in line with the most recent developments in science and technology. Moreover, he or she must also operate a quality management system that includes good manufacturing practice, in line with the type and scale of his activities. The EU guidelines for good manufacturing procedure for medicinal products must be consulted when the foundations for good manufacturing practice are laid out. This gives the EU guidelines for good manufacturing practice a high regulatory binding character. Bear in mind that methods other than those described in the EU guidelines may also be suitable for implementing the goals of the quality assurance principles. The EU guidelines for good manufacturing practice for drug products do not, in any way, intend to restrict the development of new concepts or technologies providing that these are validated and ensure a level of quality assurance that is at least equal to that described in the EU guidelines. The EU guidelines, therefore, have the character of prefabricated expertise representing modern scientific and technological standards for drug product manufacturing and testing.
In accordance with article 10 para. 3 of Directive 2003/94/EC, it is incumbent upon all European manufacturers to validate new manufacturing procedure and significant changes.
The procedures applied in manufacturing must be validated in line with the most modern scientific and technological standards. Critical phases in a manufacturing procedure must be revalidated on a regular basis. When test preparations are used, the manufacturing process must be validated as a whole as far as this is indicated, and the production development phase must be allowed for; critical processing steps must always be validated. All steps taken for the design and development of the manufacturing process must be documented in full.
1.2 Responsibilities
In Europe, the head of production is responsible for validating the manufacturing area. In accordance with §§ 2.5 and 2.7 EU GMP Guide, he or she must ensure that the necessary manufacturing procedure validations are carried out. The written procedures and operating procedures (batch production records) for which he or she is responsible for creating form the basis for validations, and must conform to the marketing authorisation/registration documents. If other internal areas (e.g. Engineering, Research & Development) are involved in the validation, responsibilities should be clearly defined.
If the task of process validation is transferred to third parties, a written contract must be drawn up between the contract giver and the contract acceptor, in accordance with EU GMP Guide § 7.1. The contract must clearly define the responsibilities of both sides and, in particular, regulate compliance with good manufacturing practice. The contract giver must ensure that the contract acceptor carries out the task in line with the instructions given. Transferring the task of process validation to external service providers does not change the regulations concerning responsibility in line with EU GMP Guide in any way: the head of manufacturing can transfer the execution of, but not the responsibility for process validation. This means that he or she retains the legal and public responsibility for completing all validation work in line with regulations in his or her area.
The holder of the manufacturing authorisation is responsible for ensuring that function owners under public law are able to carry out their duties in accordance with the regulations. In accordance with EU GMP Guide § 2.2, he or she must bestow sufficient authority on staff in leading or responsible roles to enable them to meet the demands of their tasks. He or she must, therefore, make the necessary organisational arrangements (organisational diagrams and job descriptions) and provide the necessary utilities.
1.3 GMP Requirements
Detailed regulations on the aims and execution of process validation can be found in the EU guidelines for good manufacturing practice for medicinal products. According to chapter 5.22, when new batch production records or processing methods are introduced, proof of their suitability for routine operation should be established. It should be demonstrated that the defined process using the established materials and equipment will consistently produce a product that is of the required quality.
The process for executing validation is described in Annex 15 of the EU guidelines for good manufacturing practice for medicinal products. As Annex 15 contains only the principles of qualification and validation, the PIC/S document PI 006 Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation can assist with interpretation and implementation. This document applies primarily to inspectorates in the PIC/S member states, for whom it is intended as instructions for preparing an inspection and an advanced training aid for qualification/validation. As, for PIC/S purposes, this reflects the latest scientific and technological developments, valuable information regarding the implementation of the specifications in Annex 15 may also be found here for the industry (see chapter C.6.15 Annex 15 Final Version - Qualification and validation and chapter F.1 Recommendations on Validation Master Plan Installation and Operational Qualification Non-Sterile Process Validation Cleaning Validation (PIC/S PI 006)).
Figure 7.A-1 gives an overview of relevant text passages in the regulations.
|
Regulations relating to process validation |
|
|---|---|
|
Directive 2003/94/EC, article 10 para. 3 |
Validation of new manufacturing procedures and all important changes |
|
EU guidelines for good manufacturing practice for medicinal products, chapter 5.22 |
When new batch production records or processing methods are introduced, proof of their suitability for routine operation should be established. It should be demonstrated that, when the established materials and equipment are used, the defined process will consistently produce a product that is of the required quality. |
|
Annex 15 of EU guidelines for good manufacturing practice for medicinal products |
Description of the validation process |
|
PIC/S document PI 006 "Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation, Cleaning Validation" |
As interpretation and implementation aid for Annex 15 of the EU GMP Guideline |
1.4 Aspects regarding marketing authorisation
Process validation must thereby take into account the critical parameters that can influence product quality or process reliability. Determining critical parameters already forms part of the development and improvement phases of the process. The manufacturing methods should be founded in these phases and a description of the necessary in-process controls should be given. Information regarding the evidence required for new drug approval with respect to the development of a drug product and its relevant production processes can be found in the Note For Guidance on Pharmaceutical Development (EMEA/CHMP/ 167068/2004, the document is the same as the ICH Q8 document) and the EMEA Note For Guidance on Development Pharmaceutics (CPMP/QWP/ 155/96), among other places. In these guidelines, it is clearly emphasised that the process development studies form the basis for later validation inspections (see figure 7.A-2). In this respect, development studies carried out before marketing authorisation may also be the subject of official controls in a GMP inspection.
With regard to amendments that become necessary later on during process improvements, the applicant can ensure maximum flexibility in the future as early as during the authorisation process: he should describe the measuring equipment available for continuous process monitoring and state how the end point in subprocesses can be controlled and reviewed. If monitoring data from the development phase is summarised meaningfully, this can also be evaluated as evidence that he or she understands the process. His or her ability to control critical process parameters should also be proven by these means.
The process robustness and its ability to produce reproducible quality should be proven by means of risk evaluations, among other methods.
The applicant should submit a full description of the manufacturing procedure, on the basis of the critical product and process attributes detected in the development phase. The requirements for this are described in the EMEA Note for guidance on manufacture of the finished dosage form (CPMP/QWP/486/95). The significance of in-process controls and the procedure with regard to process optimisation should also be addressed in this description.
If certain batch-related quality controls are to be omitted, the reason for this must be provided by means of data from the process evaluation or validation. See the chapter E.13 Q6A: Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances for more information.
|
Relationship between process development and validation |
|---|
|
The selection, control and any improvements to the production process that are to be described in chapter 3.2.P.3.3 of the submission file for marketing authorisation (Common Technical Document) must be explained in further detail. The critical characteristics of the formulation, as well as the process options available should be taken into account in order to give reasons for the selection of the manufacturing procedure and the formulation components. The suitability of the equipment used for the product should be demonstrated. Process development studies should form the basis for process optimisation, process validation and continuous process verification. (In accordance with the EMEA Note For Guidance on Pharmaceutical Development, EMEA/CHMP/ 167068/2004) |
A process description and the proof that the finished product conforms to its specifications are, by themselves, no guarantees that the manufacturing process is suitable. For this reason, the applicant is increasingly expected to present data regarding the validity of the manufacturing process as early as during the authorisation process. The scope of the data to be presented depends on the complexity of the product and the relevant manufacturing process. The EMEA Note for Guidance on Process Validation (CPMP/QWP/848/96) indicates the required data volume that should routinely be presented in the authorisation process.
The applicant's job is made significantly easier by the fact that the Note for Guidance on Process Validation for marketing authorisation of the drug product does not require proof of the three validation batches on a commercial scale that are otherwise customary in validation. This also applies for all standard manufacturing procedures in which the data regarding the pilot batches (see below) allows validity on a commercial scale to be predicted. The validation on a commercial scale is then executed in the manufacturing facility after approval has been issued, and reviewed by the local GMP inspectorate.
Whenever deviations from standard manufacturing procedures are found, proof of the validation on a commercial scale must also be recorded in the application for marketing authorisation. Annex II of the Note for Guidance on Process Validation clarifies which cases are considered non-standard manufacturing procedures (see figure 7.A-3). The active substance used, type of drug product, the process itself and the manufacturer's production experience all play a part in deciding whether a manufacturing procedure is a non-standard manufacturing procedure.
|
Non-standard manufacturing procedures in accordance |
|---|
|
The Note for Guidance on Process Validation assumes that the applicant is in a position to establish a relationship between the data in the development phase (laboratory and pilot batches) and the results of later process validation on a commercial scale:
- An initial inspection of the suitability of the procedure and its in-process controls should be carried out in the earlier development phase by manufacturing batches on a laboratory scale (laboratory batches: 1/100 to 1/1000 of the later market size). The laboratory batches are usually used to develop bulk manufacturing and packaging procedures. These batches can also be used in preclinical or clinical studies. The manufacture of laboratory batches is an effective means of determining critical product and process parameters. This is the stage at which suitable reasons should be given for the selection of the manufacturing procedure.
- Following on from this, pilot batches are manufactured as part of the process improvement phase, with a batch size at least 10 % of the later commercial batches. In the case of solid oral dosage forms, the batch size must be at least 10 % or 100,000 units, whichever is larger. (Exception: in the case of veterinary medicaments, the batch size may also be below 100,000 units). The pilot batches thereby form an intermediate stage between the small scale of the laboratory batch at the development level and the large scale of the commercial batch in routine production. This intermediate stage serves to predict the feasibility of manufacturing on a commercial scale. With the pilot batches, the controllability of critical parameters in the manufacturing process should be reviewed under conditions similar to the routine. It is also important to determine which equipment is suitable for manufacturing on a commercial scale. Like laboratory batches, these batches can be used in preclinical or clinical studies. They also act as a sample for stability testing.
Figure 7.A-4 Validation diagram in accordance with Annex I of the Note for Guidance on Process Validation
Validation diagram in accordance with
Annex I of the Note for Guidance on Process Validation- Brief process description with a summary of the critical process levels or critical variables to be investigated during the validation
- Specifications of the finished product
- Cross reference to analytical methods in the submission file for marketing authorisation
- In-process controls and acceptance criteria
- Additional tests to be carried out (with acceptance criteria and evidence of validation of analytical methods)
- Sampling planning: where, when and how will the samples be taken?
- Data on the record type and assessment of results
- Time planning
- The transition from laboratory scale to pilot batch size to commercial scale (scale-up) should verify that the batch size can be enlarged without impairing product quality. To this end, a diagram of the process validation to be carried out later on a commercial scale should be submitted with the application for marketing authorisation, indicating in particular how the critical parameters detected in the development phase should be reviewed at the commercial batch level. The validation diagram should be submitted with the application for marketing authorisation and should contain, as a minimum, the information displayed in figure 7.A-4.
Once validation has been completed in line with the diagram above, a validation report should be created that contains the information listed in figure 7.A-5, as a minimum. If significant deviations from the results expected have been determined at validation, the regulatory authorities and authorities responsible must be informed immediately. In such cases, corrective action must be proposed. If this results in any changes to the manufacturing procedure being necessary, an amendment must be submitted to the regulatory authorities.
|
Validation report in accordance with |
|---|
|
2 Principles of process validation
Lack of understanding of the purpose and aim of process validation and incorrect interpretations of the regulatory basics have, in the past, overwhelmingly caused process validation to be mutated into a documentation exercise in which three validation batches are manufactured on the basis of fixed, mostly average parameters without risks and process limits being identified. The validation documentation is archived and not usually used again. This procedure cannot lead to success with regard to the process reliability that is to be demanded, and even today it no longer complies with the understanding of a proper process validation. The manufacturer should be able to prove the validity of his processes on an ongoing and permanent basis, not only at the start and at certain points in the process.
The manufacturing systems used and the equipment used for the in-process control must have been qualified conclusively before validation is executed. The personnel that conducts (i.e. controls or supervises) the manufacturing process must have been trained appropriately in the tasks to be carried out. All documents used (operating instructions, records, checklists) should be checked, approved and implemented in advance.
Relevant acceptance criteria for validation in particular should be derived from the work carried out in the research and development phase. An acceptance criterion is an established requirement that must be met for a validation to be completed successfully. Acceptance criteria can be set as both process-related parameters and product-related specifications.
Acceptance criteria must be created before validation is carried out as they are core elements of every validation protocol. When drawing up acceptance criteria, it is possible to draw on requirements from batch production records, application documentation or risk analyses, for example. Figure 7.A-6 gives an overview of the most important principles of process validation.
|
Principles of process validation |
|---|
|
2.1 Process understanding
In order to be able to validate processes, first you have to understand them. You need to know how and by what they are influenced in order to be able to control processes adequately. This kind of risk-based approach must be pursued throughout the entire lifecycle of the process, starting with development and optimisation and continuing to routine production and change control. The trend in process validation is, therefore, clearly towards a continuous process validation. This means that processes are not simply validated once and then their suitability not reviewed for a long period, but they are continually monitored and evaluated. Statistical tools such as quality control cards play an important role in this.
The work carried out in the development and improvement phase of a drug product (see chapter 7.I.1 Process development) forms the basis for process understanding. If critical parameters have already been identified in the development and improvement phase, these should be taken into account when process validation is carried out. If they are incorporated in process validation, this data is also subject to inspection by the authorities. For this reason, development and optimisation, including all changes implemented during these phases, must be carefully documented so that the compilation of the process design is traceable. Validation is consequently founded on the development and improvement phase and represents a fundamental part of the life cycle of the process (see figure 7.A-7).
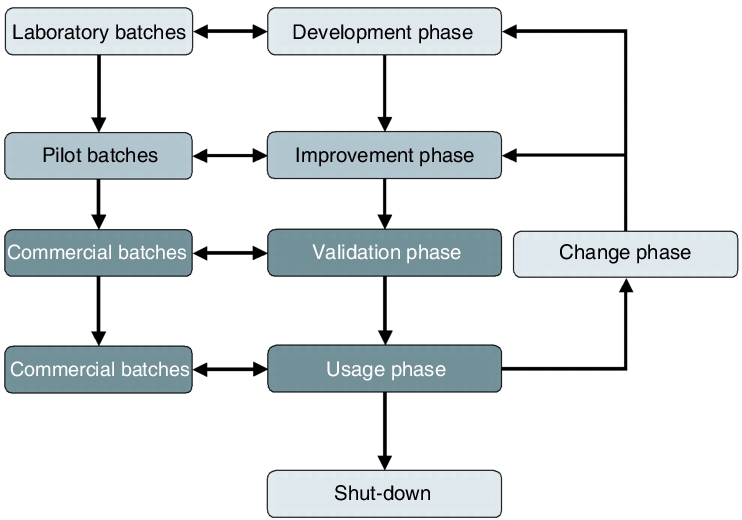 |
2.2 Type and scope of process validation
"It is impossible to state categorically when and how qualification/validation should be carried out, as the size and complexity of production processes and facilities vary considerably" (PIC/S document PI 006, chapter 2.5.5)
The expenditure required for process validation depends on the type of manufacturing procedure and the nature of the products. In the same way, a distinction should be made according to application, e.g. whether the data is to be ascertained for the approval of a drug product with a new or known active pharmaceutical ingredient, for confirmation of a change that has been carried out or for a routine revalidation.
Each process validation is based on an interpretation of a base of representative data. Validation must be carried out in such a way that the type and scope of the data received allow sufficient evidence to be provided of the reproducibility of the process. This presupposes that the basic data is complete, correct and has been recorded using calibrated measuring equipment. The interpretation always begins with the raw data upon which all processing and transfers are based.
Processes in which the interaction between critical process parameters and the variable properties of the starting material is not known exactly can only be reproduced rigidly, on the basis of established manufacturing instructions. The manufacturing instructions usually follow from the process descriptions specified in the marketing authorisation. The process description is subject to a risk analysis, the results of which determine the scope and depth of the validation (see EU GMP Guideline, chapter C.6.15 Annex 15 Final Version - Qualification and validation, no. 1). Even if process validations for standard processes (that is, all conventional, well-established manufacturing procedures apart from the non-standard processes in figure 7.A-3) are usually carried out only after approval has been granted, the prospective validation using these validation batches must still be carried out (see chapter 3.1 Prospective validation). This means that a drug product may be put into circulation only once the relevant manufacturing procedure has been confirmed as valid.
|
Statement by EMEA on continuous validation |
|
Question
Answer
(EMEA homepage, Q/A on PAT) |
|
Statement by FDA on continuous validation |
|
Advanced pharmaceutical science and engineering principles and manufacturing control technologies can provide a high level of process understanding and control capability. Use of these advanced principles and control technologies can provide a high assurance of quality by continuously monitoring, evaluating, and adjusting every batch using validated in-process measurements, tests, controls, and process endpoints. For manufacturing processes developed and controlled in such a manner, it may not be necessary for a firm to manufacture multiple conformance batches prior to initial distribution. Agency staff (field and Center) should discuss the need for conformance batches prior to distribution with the designated agency contacts when inspecting firms employing these advanced pharmaceutical science and engineering principles and control technologies. (FDA Compliance Policy Guide CPG 7132c.08, 2004) |
If, however, the manufacturer is in a position to continuously evaluate a process, e.g. by using process-analytical technologies (see chapter 7.J Process Analytical Technology (PAT)) as well as process control and evaluation by means of control cards (see chapter 6.3 Quality control cards), and can give evidence of the process understanding required for this, he or she has reached a status that can be labelled continuous validation. Where possible, this should be proven as early as in the approval procedure (see chapter 1.4 Aspects regarding marketing authorisation). In such cases, regulations can be simplified; a prospective validation using three validation batches on a commercial scale can be omitted. The concrete evidence that must be presented in individual cases for a continuous validation status to be assumed has not yet been substantiated by the authorities. Both the European (EMEA) and American pharmaceutical authorities (FDA) have, however, declared that they would, in cases where extensive process understanding has been proven, be prepared to dispense with the need for evidence of the three validation batches (see figure 7.A-8).
2.3 Traceability of validation investigations
Validation investigations should deal in particular with the critical product and process attributes that were determined during the manufacturing procedure's risk analysis. Manufacturing procedure, risk analysis and scope of validation are, therefore, very closely related.
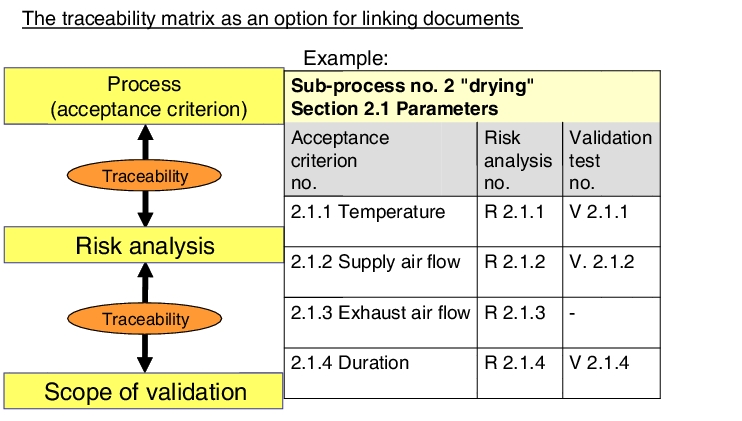 |
The type and scope of the validation investigations carried out are, therefore, traceable only if this relationship is also clear in the documentation. Criticism is often heard in GMP inspections to the effect that the risk analysis does not take the entire manufacturing process into account and the scope of validation cannot be harmonised with the results of the risk analysis. Setting up a comparative process matrix (traceability matrix) has proven useful in making it possible to prove the traceability of the validation results to the results of the risk analysis and the functionality of the manufacturing process (see figure 7.A-9).
2.4 Manufacturing under routine conditions
Process validation should be carried out under conditions that are as close as possible to reality, and should take into account the following factors, among others:
- Raw materials from different suppliers/manufacturers used, in particular in different synthesis procedures/impurity profiles of active pharmaceutical ingredients
- Validation batches manufactured by different personnel (insofar as manual influences are critical)
- Routine production equipment used (different facilities/lines should also be compared where it is relevant and critical)
- Validation batches manufactured in different market sizes (providing the batch size is a critical parameter)
- Seasonal, climatic influences taken into account (insofar as e.g. temperature and humidity may affect the product or process)
2.5 Bracketing (product group formation)
Depending on the product range and type of production, it is possible to combine groups of products or procedures (bracketing), which reduces the total expenditure required. The principle behind bracketing and the classification of individual products or procedures into one group must be justified. The validation object selected from the group does not need to be representative of the entire group, but should also represent the Worst Case. The Worst Case is usually the manufacturing procedure that is hardest to control - a selection which must be justified based on a risk analysis.
2.6 Challenge tests
Also, in the framework of process validation, proof should be established of the suitability of sampling procedures and in-process testing regarding their significance for process reliability. Usual procedures to do this are challenge tests, which are intended to prove that errors are discovered and rectified or prevented by means of existing control measures (e.g. the inspection to ascertain whether a camera-assisted blister control system is capable of determining incorrectly-filled blisters).
The aim of these challenge tests is not to determine margins of error (proven-acceptable ranges, PAR). This is a subject addressed in the development stage. The validation is executed under standard conditions, not extreme conditions that do not conform to reality. Challenge tests under worst-case conditions do not mean that a number of validation batches must be manufactured in consideration of all possible combinations of critical parameters. (If only five parameters each have three test values, this means that 35 = 243 validation batches are manufactured on a commercial scale.) This means that the standard conditions that are the least favourable for the process and product when all circumstances are taken into account are those that are selected as the result of a risk assessment.
As evidence that the specifications have been fulfilled, all required quality controls should also be carried out on the finished product .
2.7 Deviations
Each process validation must be documented completely and in a such way that it is traceable. Changes in the test procedure must be noted. If batch data has not been considered, this must be explained and documented.
Deviations during the validation (e.g. deviations from acceptance criteria, the procedure or specifications) must always be documented. The cause of the deviation must be determined and the consequences evaluated. During validation investigations, deviations can occur if the design of the process that is being checked was not suitable, or the process was not optimised sufficiently with respect to the control parameters. Inadequate process development or optimisation must be rectified later. This subsequent improvement must not, however, be made by means of the validation since it is not the aim of validation to develop or improve, but to provide evidence that the product is, since development and optimisation, suitable in accordance with previously specified acceptance criteria. It follows that it is prohibited to make any alteration to a previously established sequence of operation or acceptance criteria in the context of finalising a validation protocol. In these cases, the optimisation of the process should first be halted, appropriate change control procedures carried out and validation begun.
3 Types of validation
3.1 Prospective validation
In principle, it is assumed that process validation is completed before the first routine manufacture of the drug product (prospective validation). In the case of standard manufacturing procedures, this takes place after approval has been granted; for non-standard manufacturing procedures, validation must already have been proven in the approval procedure (see chapter 1.4 Aspects regarding marketing authorisation).
To confirm that a prospective validation has been carried out in accordance with the regulations, the requirement is for three consecutive batches conforming to specification to be manufactured. The batch size should correspond to the subsequent commercial scale. The manufacturer must justify testing on smaller validation batches and simultaneously provide evidence that the results can be transposed to the commercial scale. If this is the case, it should be justified in detail in the validation protocol. Finally, evidence of the validity of the process must be recorded even if batch size is reduced.
The validation batches manufactured may only be put into circulation if a positive validation result has been recorded. Similarly, no commercial batches may be put into circulation until validation has been successfully completed. Since stock for sales or for other forms of disposal meets the conditions of placing on the market, it is permissible to begin routine manufacturing for pharmaceutical purposes only once validation has been successfully completed (figure 7.A-10).
All requirements established in the submission file for marketing authorisation must, of course, be met. Prospective validation should be documented by a validation protocol and a validation report.
|
Prospective validation in accordance with Annex 15 of the EU GMP guidelines |
|---|
|
The points listed in figure 7.A-10 should be the minimum contents of a prospective validation should be included as points listed, in accordance with Annex 15 of the EU guidelines for good manufacturing practice for drug products (chapter C.6.15 Annex 15 Final Version - Qualification and validation)
It is generally acknowledged that the three prospective validation batches that are usually accepted have little statistical significance regarding process reliability. It is not, therefore, the rigid reproduction of three batches that is decisive within the framework of prospective validation. The number of times a process is run should rather be sufficient for considering variability in routine production, showing possible trends and obtaining sufficient data for a meaningful interpretation.
3.2 Concurrent validation
When manufacturing procedures are validated, the prospective course of action is the norm. In exceptional cases, validation of the process during routine manufacture may be required (concurrent validation). The significant difference here is the option of putting validation batches into circulation even though validation is not complete and no conclusive evidence exists that the process is indeed suitable. This represents a risk for drug product safety and consumer health protection. For this reason, concurrent validation must be used only in justified exceptional cases. The decision to take this course of action must be well-founded, documented and approved by an authorised person.
An important condition for concurrent validation is that the process is already well managed. Evidence may, among other methods, be provided by means of quality control cards (see chapter 6.3 Quality control cards) and statistical investigations into relevant process parameters, e.g. by determining the process capability index (CpK, see chapter 6.4 Process capability investigation). Development and optimisation data, data from the scale-up phase or comparable production data from other plants, for instance, may be used as data sources.
Prerequisites for concurrent validation include:
- The premises and equipment used for the process are conclusively qualified.
- A carefully-conducted risk analysis has been presented and evaluated.
- The execution of concurrent validation is described in a validation protocol, which takes into account the critical parameters determined in the risk analysis and determines acceptance criteria.
If these prerequisites are met, concurrent validation is permissible, e.g. in the following cases:
- Transfer of a validated process to another plant (e.g. to a contract manufacturer)
- Modification of an existing process (e.g. dosage or tablet shape are different from the validated procedure)
Concurrent validation is prohibited for all non-standard manufacturing procedures (see figure 7.A-3).
3.3 Retrospective validation
Processes that already exist that have not been validated prospectively or concurrently, can, in accordance with Annex 15 of the EU GMP guidelines, be validated retrospectively on the basis of historical manufacturing data (retrospective validation). Validation of manufacturing procedures has been a legal requirement for many years. In the 21st century, it is hardly imaginable or acceptable for a manufacturer not to have validated an existing manufacturing procedure. For this reason, the principle of retrospective validation can largely be revoked. Nevertheless, if manufacturers decide to use this procedure, they are advised to coordinate it with the supervisory authorities responsible.
If retrospective validation is carried out, the data must enable process reliability to be evaluated within defined acceptance criteria. Reliable data from a defined time period should be evaluated using an experience report to determine whether the manufacturing procedure in question has fulfilled the established requirements based on a validation protocol, and whether it will fulfil these requirements in future (see figure 7.A-11).
|
Data sources for an experience report for retrospective validation |
|---|
|
Annex 15 of the EU guidelines for good manufacturing practice for drug products allows retrospective validation only for established processes that have not undergone any critical changes (e.g. product composition, process parameters, process sequence) during the period under observation .
Retrospective validation is then only acceptable if the set of data used as a basis is sufficiently large (at least ten batches that conform to specifications) and statistically meaningful. A validation protocol and a validation report should be compiled for the retrospective process validation documentation, in the same way as for prospective process validation.
A purely retrospective data analysis is prohibited if:
- The raw data used as a basis is missing
- The data does not cover the current range of operational parameters
- Significant changes have been made in the period of observation
- Unresolved deviations or trends have been documented
4 Revalidation
Periodic revalidation (i.e. repetition of validation at certain time intervals) is prescribed by German pharmaceutical law: "Critical phases in a manufacturing operation must be revalidated on a regular basis" (AMWHV § 13 para. 5 clause 2). Phases in a manufacturing procedure that should be classified as critical include in particular those that may affect the product safety, such as sterilisation.
The following is also recorded in the PIC/S document PI 006 that deals with recommendations for validation: "The qualification and validation do not consist of one-off activities such as the introduction of a new manufacturing process. The initial implementation should always be pursued as a continuous programme".(PIC/S document PI 006, chapter 2.5.12)
The validation status of a manufacturing procedure can be jeopardised by the following, for example:
- Accumulation of minor changes having a negative overall effect
- Process instructions not followed correctly
- Inadequate training of personnel
- Unclear/contradictory instructions
- Change of general conditions (e.g. laws, GMP guidelines)
Manufacturing procedures should therefore be evaluated at specific intervals to ensure that they remain in a validated state. If no significant changes have been made to the manufacturing procedure, a check that provides evidence that the procedure still satisfies the prescribed requirements fulfils the necessity of a revalidation (review). This check can be made using quality control cards, process capability investigations, trend analyses or Product Quality Reviews (see figure 7.A-12), among other methods (see also chapter 15.F Annual product review).
|
Product quality reviews as an element of revalidation |
|---|
|
"Regular periodic or rolling quality reviews of all licensed medicinal products, including export only products, should be conducted with the objective of verifying the consistency of the existing process, the appropriateness of current specifications for both starting materials and finished product to highlight any trends and to identify product and process improvements." (EU GMP Guideline, Chapter 1.5). |
The schedule for revalidation should be given in a validation master plan (VMP, see chapter 5.1 Validation master plan)..
|
Changes which may make a revalidation necessary: |
|---|
|
In addition to the periodic validation, a revalidation must be carried out for critical changes too. According to Annex 15 of the EU guidelines for good manufacturing practice for drug products, it is incumbent upon the manufacturer, as part of his change control program (see chapter 19.C Change control) to evaluate all changes with regard to their effects on product quality or process reliability and carry out a revalidation if necessary.
Figure 7.A-13 mentions some examples of changes for which revalidation is compulsory.
The revalidation of critical changes should provide evidence that changes to a process and/or the process environment do not negatively affect process attributes and product quality (cf. PIC/S document PI 006, chapter 6.6.1).
It is not absolutely necessary to requalify a process from scratch just because a specific aspect has been modified. However, it is important to carefully assess the type of change (risk analysis) in order to identify possible repercussions and to establish the precise scope of the revalidation (cf. Guideline on General Principles of Process Validation, FDA, CDER, 1987 see chapter D.2 Guideline on General Principles of Process Validation).
The documentation requirements for revalidation are the same as those for initial validation and similar documents may, therefore, be used in many cases (cf. PIC/S document PI 006, Chapter 6.6.3).
5 Process validation documentation
The main documents for validation are the validation master plan and the related validation protocols with the closing validation reports.
The GMP documentation requirements (see chapter 15.B GMP-conforming documentation) apply, in general, to the design and compilation of documents for validation.
5.1 Validation master plan
A unit's current validation projects must, in accordance with Annex 15 of the EU guidelines for good manufacturing practice for drug products, be described in a validation master plan (see figure 7.A-14).
|
Contents of the validation master plan (VMP) |
|---|
|
The manufacturer can use this to establish the principles and procedure relevant to the process validation and estimate the resources required. In this respect, the timing and sequence of the individual validation projects is an important component within the validation master plan. Moreover, the validation master plan enables the GMP investigator to understand the company approach towards process validation as well as towards determining and organising the required activities. When the validation master plan is compiled, reference may be made to existing documents. In the case of large projects, it is possible and permissible to compile several validation master plans.
5.2 Validation protocol and report
The detailed rules for the validations should be established in directions based on procedure ("validation protocol" according to Annex 15, no. 6) and checked and authorised by the persons responsible. In terms of their content, they should, in particular, specify the critical steps and state the acceptance criteria.
A validation protocol should contain the points shown in figure 7.A-15 in accordance with PIC/S document PI 006, chapter 6.3.3.
|
Contents of the validation protocol in accordance with PIC/S document PI 006 |
|---|
|
The execution of individual validation projects should be documented in a report that corresponds to the validation protocol ("validation report" in accordance with Annex 15, no. 7, see figure 7.A-16), in line with specifications.
|
Contents of the validation report in accordance with PIC/S document PI 006 |
|---|
|
The report should contain an overview of the results cross-referenced with the validation protocol. Deviations observed and the conclusions drawn from them (including necessary changes) should be listed. Deviations from the plan should also be evaluated.
Recommendations for continuing investigations (e.g. trend analyses, monitoring) and the in-process controls necessary for routine production should accompany the evaluation of the report.
5.3 Archiving
According to national law all validation records must be retained in full. These archiving regulations may be transferred to validation documentation; the archiving period is calculated from the last batch manufactured using the validated procedure. Records must be archived in a suitable area within the premises established by the authorisation in line with national law. Suitable measures must be taken to restrict access authorisation to the records to authorised persons only. If the manufacturing company or testing operation in which the documentation is stored is shut down, the pharmaceutical manufacturer must take measures to ensure that the documentation is archived for the full period required.
All raw data accrued in connection with validation should be archived together with the validation report or as an annex to it. In no circumstances may raw data be destroyed before the archiving period has expired. Original documents must be stored. Summaries of raw data, e.g. in Excel tables, cannot be viewed as a replacement. In exceptional cases, copies may be archived instead of original documents if legibility cannot otherwise be guaranteed for the duration of the archiving period (e.g. if data is printed on thermal paper).
If raw data is accrued electronically, it is particularly important to consider creating legible printouts after several years have passed. This may mean that the data format or storage medium has to be changed during the archiving period. Transferring data to a new media or a different format must not alter the raw data in any way. The authenticity of the data once it has been transferred must, therefore, be ensured by means of a validated procedure.
6 Maintaining the validation status
6.1 General conditions and prerequisites
Classic approaches to validation, whether they are carried out prospectively, concurrently or retrospectively, all have the same shortcoming: they give only a snapshot of the validation status of a procedure/process. For reasons of drug product safety, however, it is important to demand that the suitability of procedures and processes be ensured on a permanent and continuous basis. The manufacturer should, therefore, take all the necessary measures to ensure that the suitability of his processes is continuously reviewed, confirmed and verified (continuous validation/verification).
Basic principles for suitable processes include:
- Established control measures
- An understanding of the connection between critical process parameters and product quality attributes
- Qualified and continuously-maintained premises, equipment and supply systems for process operation
- Qualified personnel, trained in process control and documentation
- Effective change control programs that establish and monitor the measures required for implementing critical process changes
- Self-inspections to review the effectiveness of the quality assurance system
- A system to deal with deviations
Measuring systems in particular are critical elements of the equipment required for suitable data analysis. If such systems display a high spread in relation to the overall process spread, this could cause inappropriate decisions to be made. Measuring systems must, therefore, be stable and not contribute significantly to overall data spread. They must, of course, deliver correct results, which should be ensured by carrying out calibration on a regular basis. In classic manufacturing processes, the suitability of which is confirmed predominantly by means of conclusions from random sampling on intermediate and final products, data recording from a specific time period can only be evaluated retrospectively. Process measures are, however, then only efficient and effective if they are used to prevent faults and deviations, i.e. the aim of these measures is for critical process and product attributes to conform to the specified acceptance criteria. This keeps the spread of finished products within the tolerance limits. In the case of processes for which a Design Space (chapter 7.I.2 Design space) has been described and which are monitored by means of process-analytical technologies (see chapter 7.J Process Analytical Technology (PAT)), the manufacturer continuously receives data from which he can directly derive the suitability of the process.
6.2 Principles of statistical process control
Statistical process control, in which statistical procedures are used for planning and evaluation, helps control and steer production processes (cf. DIN 58936 Quality management in laboratory medicine - Part 1: Basic terminology).
Product batches and production processes are never exactly the same, as every product and process is exposed to different influences, which cause varying measurement values for the quality attributes in question. Under spread, different measured values are summarised in the descriptive statistics to estimate the dispersion of sample values around their mean.
The following measured values are used for the spread:
- Spread
- (Inter) quartile interval
- Average deviation
- Average absolute deviation
- Variance and standard deviation
The causes of spreads that can be determined relating to one feature by observing processes may be manifold. Random and systematic influences must also be considered:
- Random influences represent the constant total of many small individual influences. The are always present, stable over time and, therefore, predictable.
- Systematic influences have a non-random, frequently consistent cause. This may be a missing verification, for instance, replaced components, external influences, ageing or fatigue. They often change the measuring signal in the same direction only. Systematic influences can be traced back to a large main influence or a few influences that occur irregularly and, therefore, render the process unstable and unpredictable. In rare cases, this changes the process to such an extent that it benefits the process products. Then, the systematic influences can be identified and introduced in the long term. As a rule, however, systematic influences are undesirable, as they change the process distribution so significantly that the products no longer conform to specifications. Harmful systematic influences must, therefore, be identified and rectified.
Processes that display a stable and repeatable distribution and are, as a result, free of troublesome systematic influences, are also known as managed processes (ee figure 7.A-17).
|
Managed process |
|---|
|
A process in which the parameters do not change the distribution of the characteristic process values, or the extent to which they are changed is known or within known limits. |
The two upper examples in figure 7.A-18 show an unmanaged process in which the cause of spread can be attributed to both random and systematic influences. Providing that the project range remains within the tolerance limits as shown in the second example, products that conform to specifications will still be produced. However, as the process is not managed, the result of even the next batch cannot be predicted, and the process conditions may breach the tolerance limits. In both of these cases, systematic spread influences must first be eliminated so that the process is manageable.
Both of the lower examples in figure 7.A-18 show processes in which conditions remain largely unchanged, which can be attributed to constant statistical characteristic values (mean, standard deviation, range). The managed process that does not, however, conform to specifications (third example in figure 7.A-18) still needs to be optimised (to reduce random spread influences) to ensure that all units conform to specifications.
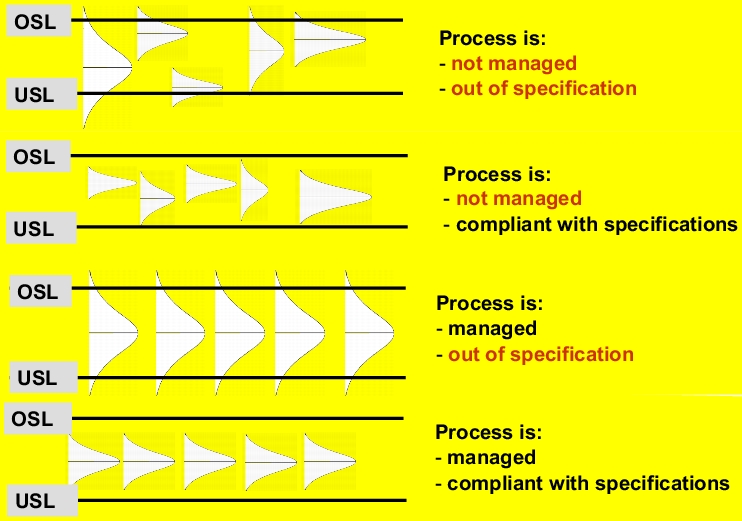 |
.
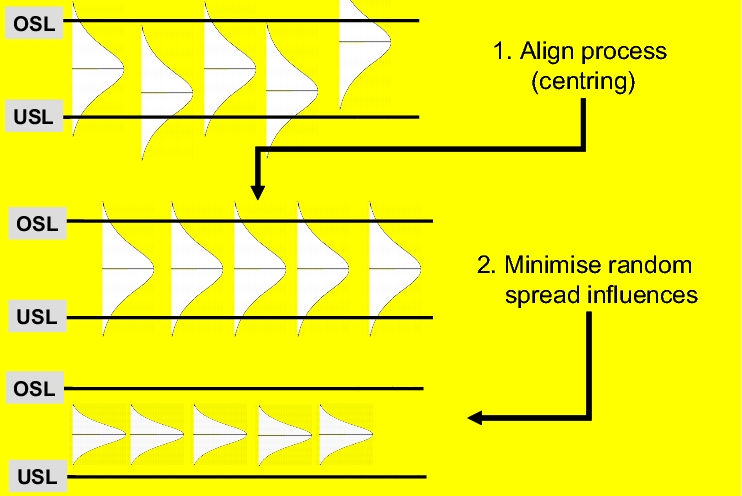
|
The aim of statistical process control is to control a process in such a way that systematic influences are excluded and the process adheres to the established acceptance criteria.
Figure 7.A-19 also reflects the procedure in process control:
- The first measure in process control is aimed at aligning the process with a standard value (centring it).
- In order to reduce the proportion of products above and below the specification limit, random spread influences must also be reduced (second measure). This increases the yield of products that conform to specifications.
- An acceptable (capable) process is characterised by the fact that its internal spread is lower than the specified tolerances. In the example diagram, the tolerance conforms to the interval between the upper specification limit (USL) and lower specification limit (LSL).
Since a managed process can generally be described by means of a predictable distribution, this distribution can be used to estimate the number of products that conform to specifications. Providing that the statistical characteristic values of a process do not change significantly, i.e. the process is managed, it can be assumed that it will generate a consistent number of products that conform to specifications
6.3 Quality control cards
Dr. Walter Shewhart distinguished between process data that is controlled and uncontrolled, that is between processes subject to random and systematic influences, as early as 1931 in his book Economic Control of Quality of Manufactured Product. Shewhart used a graph, the quality control card, to develop a way of identifying and counterbalancing changes in the process flow at an early stage, using sampling data. The testing data from the random sample, such as tablet weight, is shown in graph form on a quality control card QCC (see figure 7.A-20).
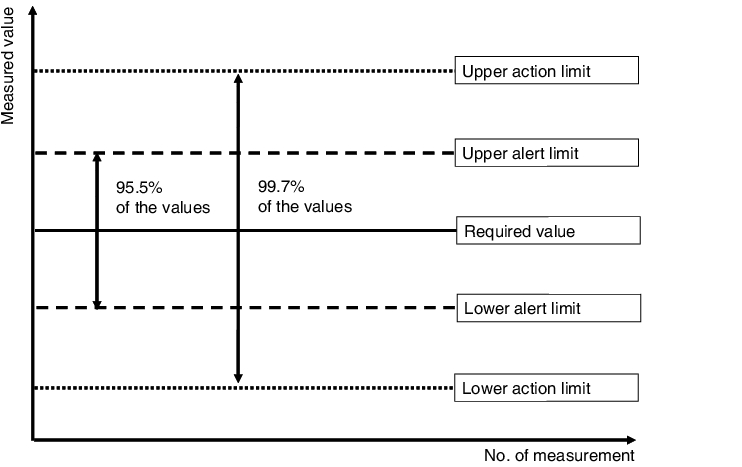 |
The alert and action limits plotted on the quality control cards indicate the values at which an early warning of a possible deviation from normal operational parameters could occur and immediate corrective action and further clarification is required.
Alert and action limits allow any development towards defective products to be identified in good time, before a faulty part is produced. This enables you to intervene in the process early enough to prevent defective products from being produced. The quality control card is, therefore, an indicator of a process's ability to supply products that conform to specifications. Quality control cards can be set up for both quantitative (measurable variables) and qualitative features (attributive properties) (see figure 7.A-21).
|
Control cards for quantitative features |
Control cards for qualitative features |
|---|---|
|
|
Preliminary investigations
Before a quality control card investigation can begin, the uncontrolled process sequence must be watched in an observation phase, by means of random samples (usually 10 random samples of 5 parts each). This should give a picture of the process spread behaviour. Normally-distributed values display a symmetrical distribution pattern on the quality control card, with the values of the random variables concentrated in the middle of the distribution and occur more and more sparingly with greater distance to the middle. As can be seen in figure 7.A-22, 95.5 to 99.7% of all values are concentrated around the middle value within a limit of 2 to 3 standard deviations. Consequently, in a managed process, very few values should be found outside these limits. These limits can, therefore, be regarded as alert or action limits.
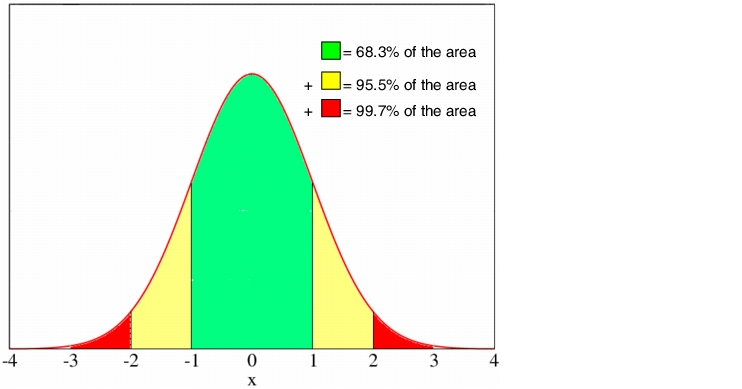 |
To be able to construct the quality control card, the statistical identification data mean and standard deviation is required. This data is determined in preliminary investigations. The mean is plotted with the relevant 2s and 3s values on the Y-axis (dimension of the feature result). The 2s values are known as alert limits and the 3s values as action or intervention limits. If the alert limit is exceeded once, this is tolerated, while if the action or intervention limit is exceeded, this requires immediate corrective action. The testing results are entered on the X-axis in chronological order. The probability of the action limit being exceeded is 0.3 %, which means that should it occur, this can safely be regarded as a deviation.
Control card types
A quality control card can be compiled for every statistical parameter that can be used to describe a process. In practice, the following types of quality control cards have become accepted:
- Mean card (mean of n units in a random sample): monitors the course of middle process conditions
- Standard deviation card (standard deviation taken from n units in a random sample): monitors the process spread
- Control cards for individual values (original values) and ranges (extreme values): uses the smallest and largest value in the random sample n in order to represent infringements of the alert and action limits
- Median control cards: an alternative to mean cards
The procedure for specifying limit values can be used to distinguish between the process control card the acceptance control card:
- The process control card is a control card that does not assume specified limits. The upper and lower alert limit as well as the upper and lower action limit are defined by means of estimated values or distribution parameters for known and previously-executed processes. Since further testing data may be gathered during the course of the process, this is used for new limit value calculations.
- The acceptance control card is a control card that is used to calculate the action and alert limits by means of specified tolerance limit values. The tolerance limit values indicate the maximum deviations that are permissible in a product.
Sign for error occurrence
The intervention limits are not the only sign that an error has occurred; the arrangement of the measuring points can also indicate this. As previously mentioned, systematic deviations are subject to principles. These principles can be deduced from the course of the measuring points on the quality control card.
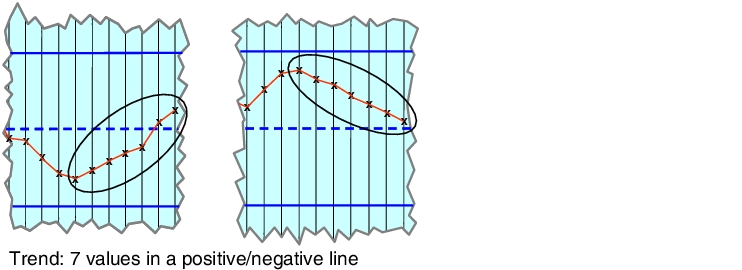
This is what is known as a trend (see figure 7.A-23), when 7 measuring points display a practically linear incline in the direction of a limit. Tool wear may be rapidly increasing, which would soon cause the intervention or alert limit to be exceeded.
 |
A pattern (see figure 7.A-24) is a non-random curve progression, e.g. periodic "oscillation" around the specified mean. This could be due to temperature fluctuations, for instance, which cause the parts manufactured to be sometimes larger, sometimes smaller.
 |
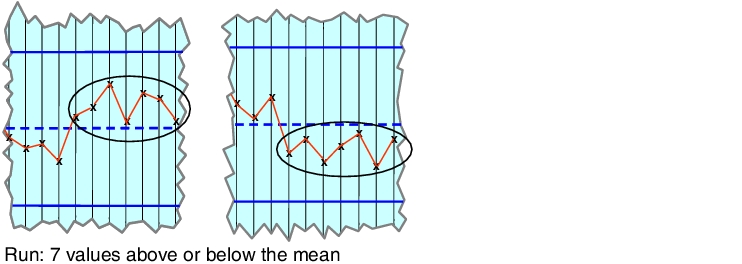 |
If 7 measuring points fall above or below the specified mean, this, for all intents and purposes, gives rise to a new real mean. This is described as a run (see figure 7.A-25). This may indicate that a stamp in a tablet press has become damaged and the pellet will, from now on, manufacture larger or smaller products.
When a quality control card is evaluated, it is essential to distinguish between random and systematic influences. Random influences cause a spread of testing data on the quality control card. They are caused by influencing factors such as temperature fluctuations or the properties of starting materials. Systematic influences lead to a gradual shift in the testing data on the quality control card, and are caused by influencing factors such as plant wear or incorrectly-adjusted equipment.
Optimum use
To make the best use of quality control cards, it is important to first clarify which issues/problems are to be dealt with by the quality control card. Understanding possible spread influences is also of some importance here. As a rule, quality control cards compare the spread within a random sample with the spread between more than one random sample. It is important, therefore, for the random samples to be taken in such a way that allows as many spread influences and sources that could affect process results as possible to be taken into account. Furthermore, it is very important to record types of measuring data that allow a real statement about product quality or process manageability and can be obtained quickly by measuring, so that the process can be controlled immediately.
6.4 Process capability investigation
Process validation must verify that the critical parameters of a manufacturing procedure or a group of manufacturing procedures are managed within established limits (acceptance criteria). This can be done by statistical investigation of the relevant process parameters on the basis of manufacturing data, e.g. by determining the process capability (process capability index (CpK)), see figure 7.A-26).
This is of particular importance in the case of processes that are complex or hard to control. If such processes are evaluated regularly by means of process capability investigations, this allows a lasting conclusion regarding validity to be drawn.
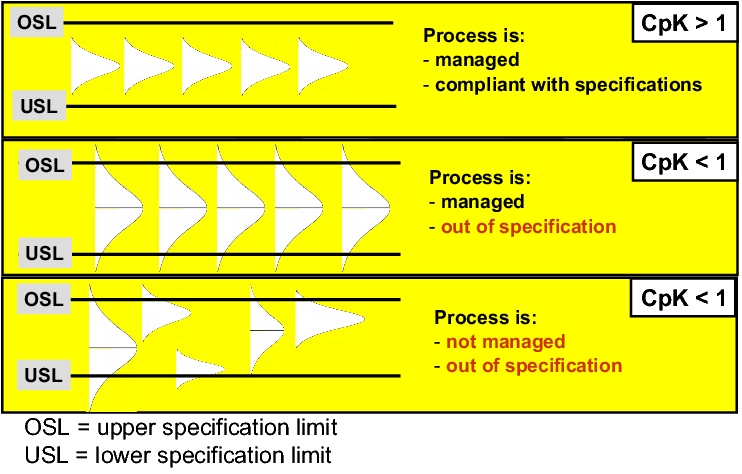 |
Managed processes with conditions that change only within tolerance limits are labelled "capable" processes. The degree to which a managed process is able to adhere to tolerance limits can also be determined by means of process capability investigations. Process capability investigations are only useful, however, if the process has previously been proven to be managed. Managed processes are predictable processes. Providing that systematic spread influences can still affect the spread and location of process distribution, calculating process capability serves no purpose. Process capability investigations also require the processes to be investigated to have a normal distribution.
Cp and CpK value
The Cp and CpK values have, in practice, become established for determining process capability
The process capability index Cp is a measure of the smallest possible proportion of defective units in a process arising when the characteristic values (centred manufacturing) are optimally distributed. The index does not state whether the distribution of the values is, in fact, centred.
The Cp value is defined as:
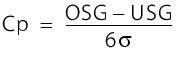
Since it is not essential for the mean value of a process parameter to conform to the middle of the specification breadth, it is more useful to determine the CpK value. The process capability index (CpK) is a measure of the proportion of units in a process that can be expected to be defective. The larger the index, the smaller this proportion.
The CpK is defined from the mean, the corresponding standard deviation and the upper or lower specification limit (USL; LSL) as follows:
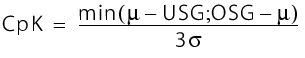
The higher this value, the more safely all units from the quantity investigated are located within the specifications.
While the Cp value indicates only the relationship of the process spread to the specified tolerance, the CpK value also includes the location of the mean in relation to the middle of the tolerance specified.
The CpK value is, therefore, always the same or smaller than the Cp value
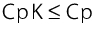
If CpK = Cp, the mean of the quality attributes (process conditions) is exactly in the middle of the tolerance. The smaller CpK is in relation to Cp, the further the process conditions are from the middle of the tolerance. If the CpK value > 1.33, process capability can be assumed (see figure 7.A-27)
|
Process capability in accordance with DIN 55350 |
|
|---|---|
|
CpK < 1.0 |
No process capability |
|
1.0 < CpK < 1.33 |
Limited process capability |
|
CpK > 1.33 |
Process capability |
|
Summary
|
